

Genetics of glioblastoma: a window into its imaging and histopathologic variability
- PMID: 21997991
- PMCID: PMC5410960
- DOI: 10.1148/rg.316115512
Genetics of glioblastoma: a window into its imaging and histopathologic variability
Abstract
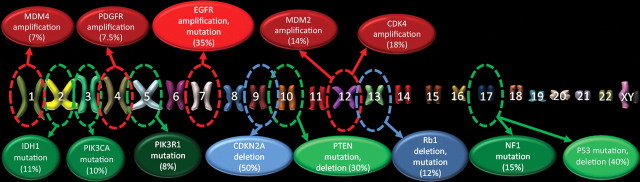
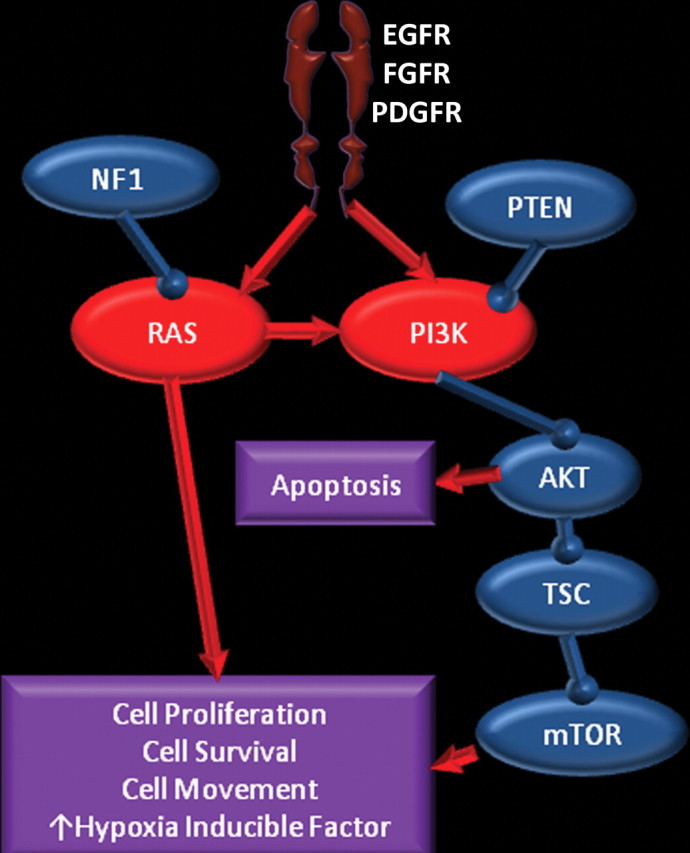
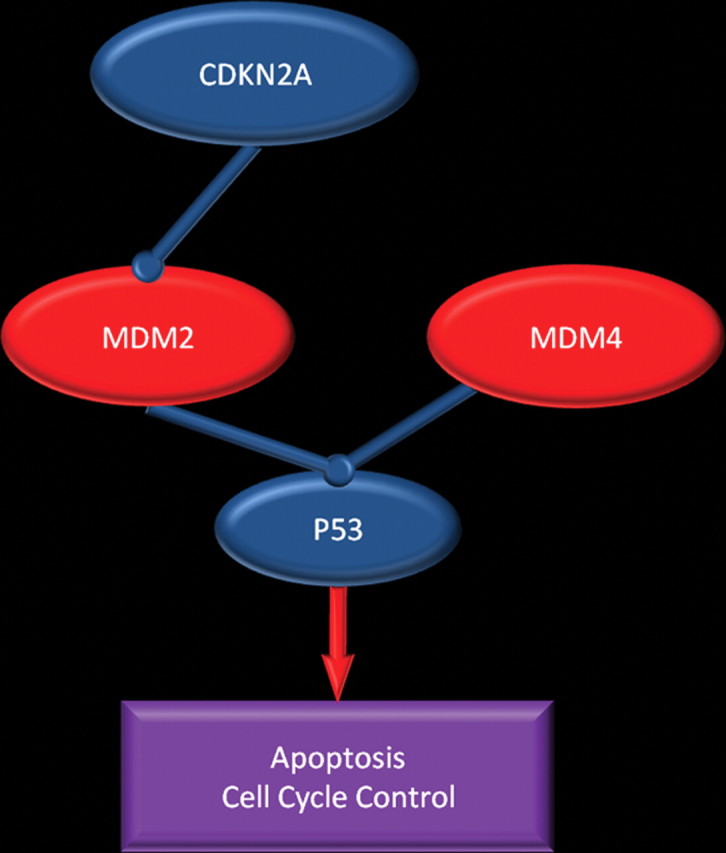
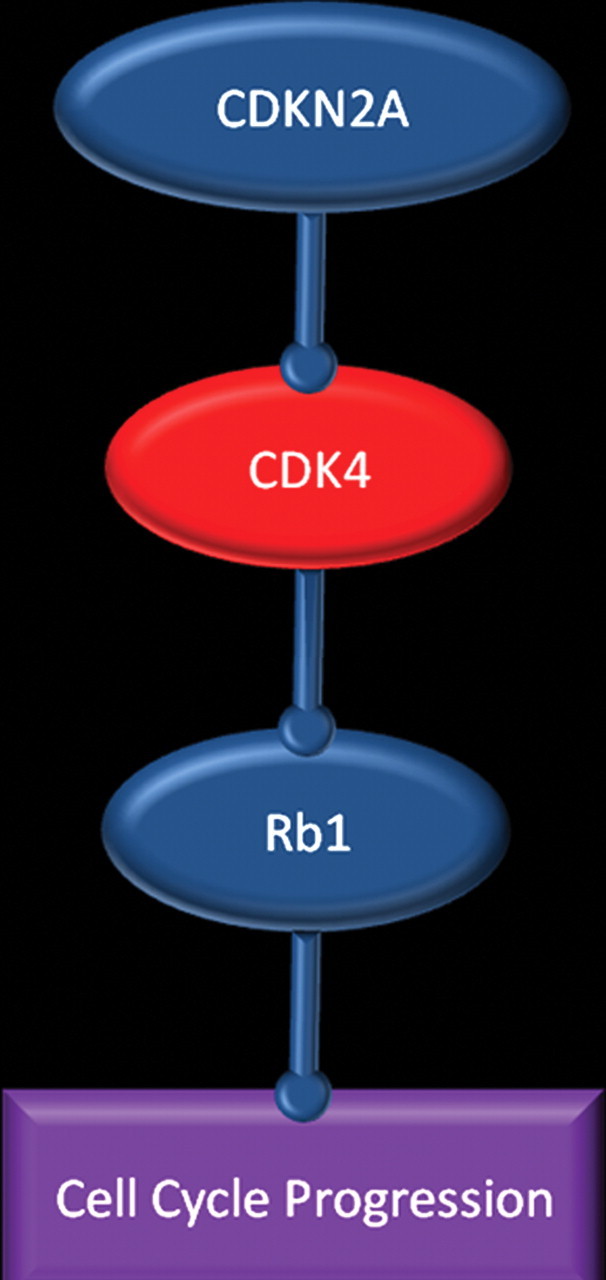
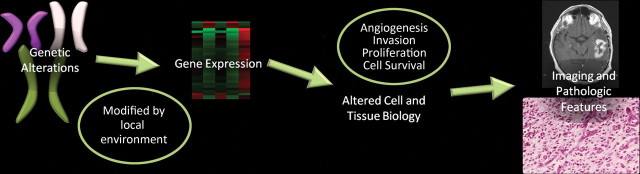
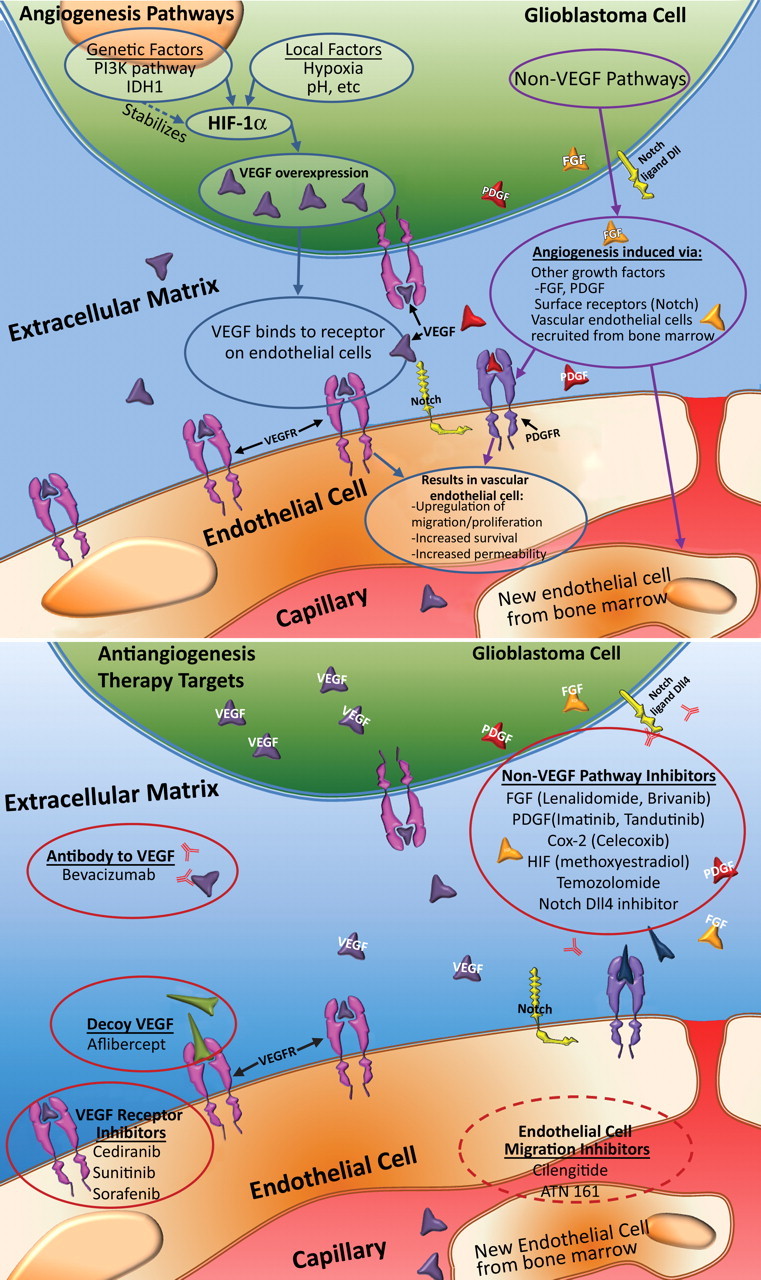
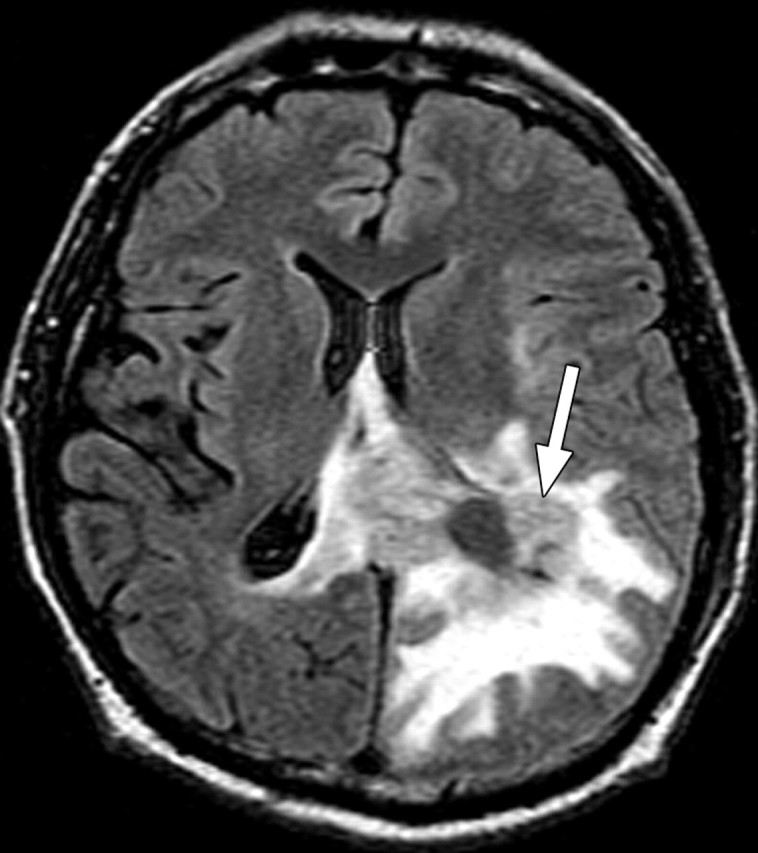
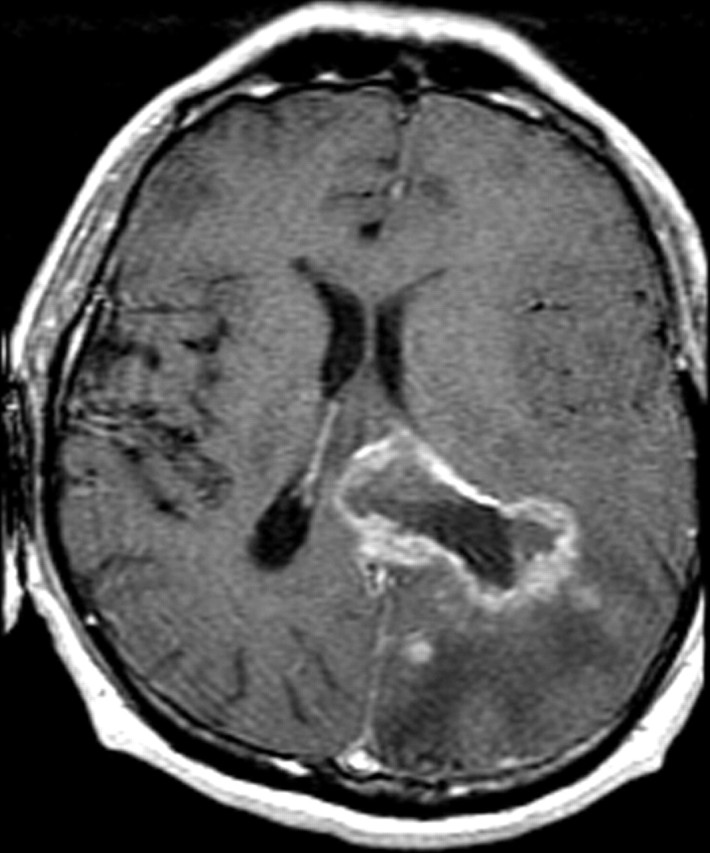
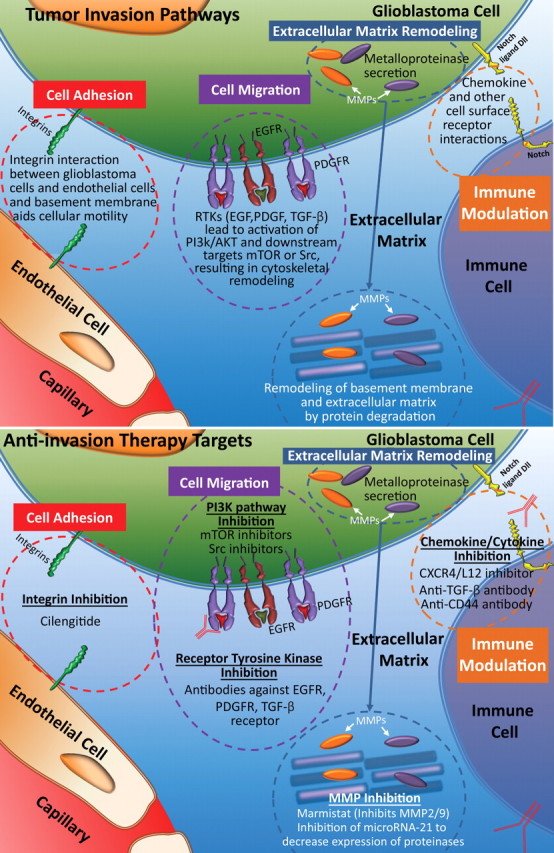
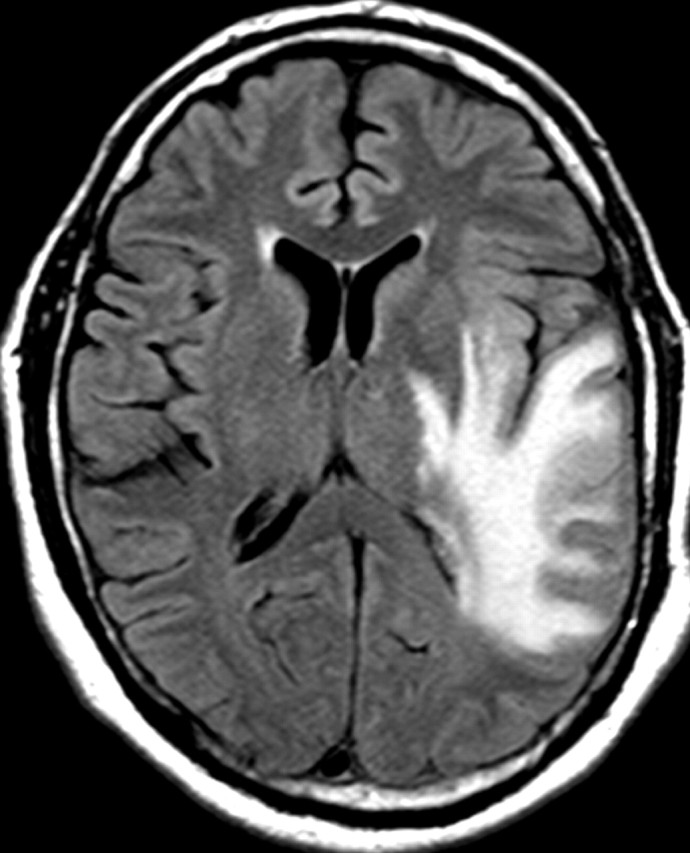
Glioblastoma is a highly malignant brain tumor that relentlessly defies therapy. Efforts over the past decade have begun to tease out the biochemical details that lead to its aggressive behavior and poor prognosis. There is hope that this new understanding will lead to improved treatment strategies for patients with glioblastoma, in the form of targeted, molecularly based therapies that are individualized to specific changes in individual tumors. However, these new therapies have the potential to fundamentally alter the biologic behavior of glioblastoma and, as a result, its imaging appearance. Knowledge about common genetic alterations and the resultant cellular and tissue changes (ie, induced angiogenesis and abnormal cell survival, proliferation, and invasion) in glioblastomas is important as a basis for understanding imaging findings before treatment. It is equally critical that radiologists understand which genetic pathway is targeted by each specific therapeutic agent or class of agents in order to accurately interpret changes in the imaging appearances of treated tumors.
© RSNA, 2011.
Conflict of interest statement
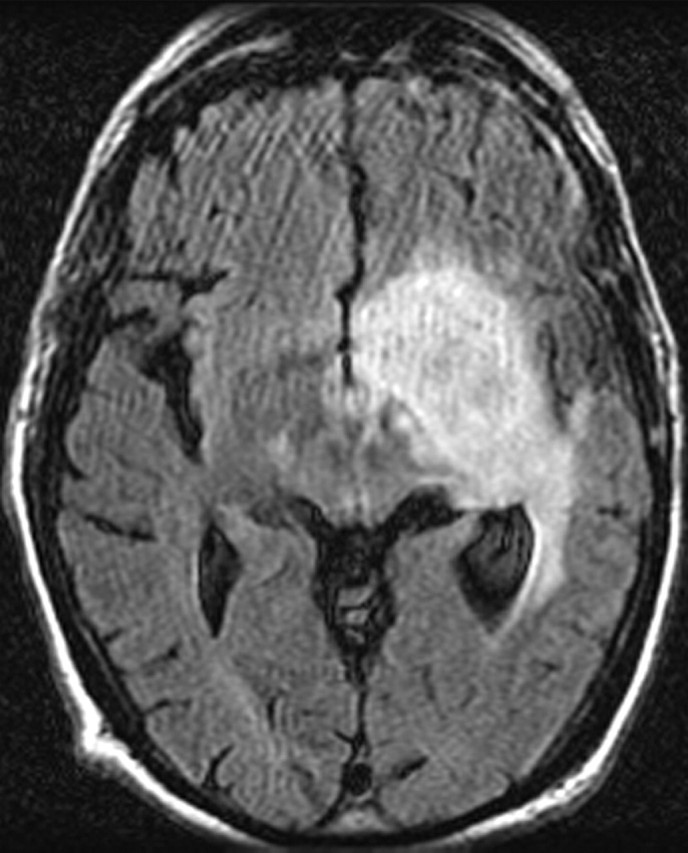
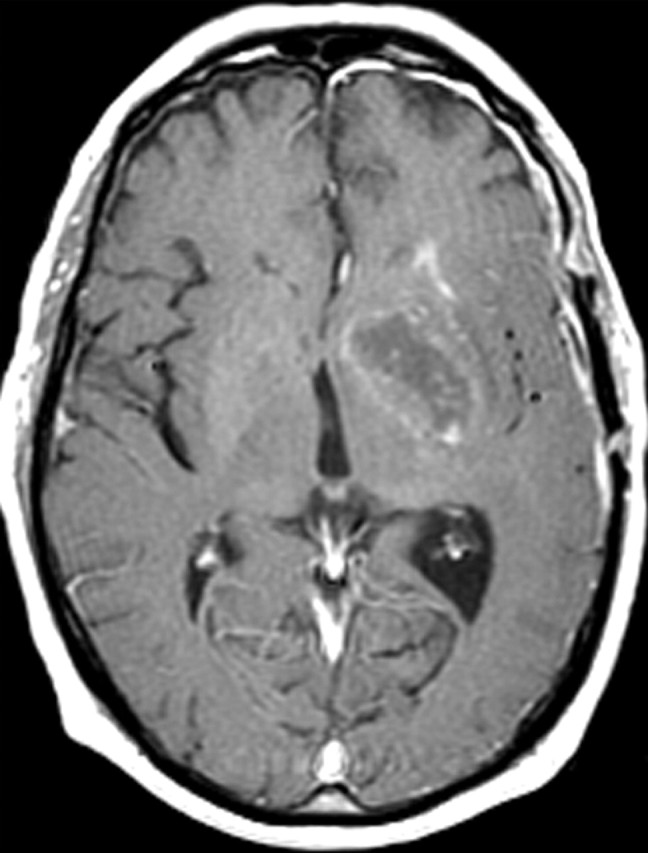
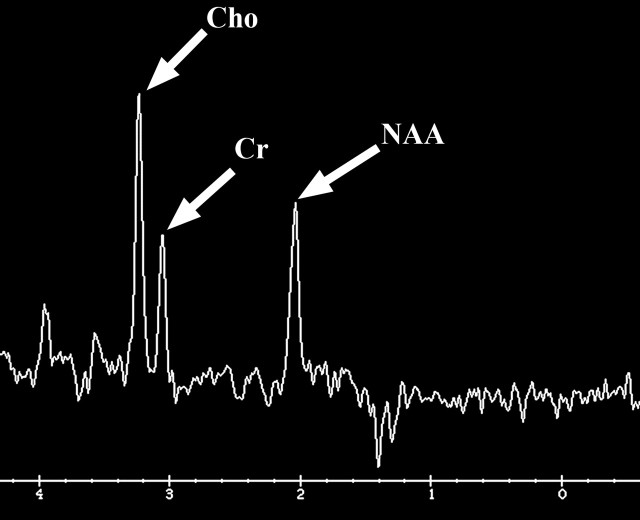
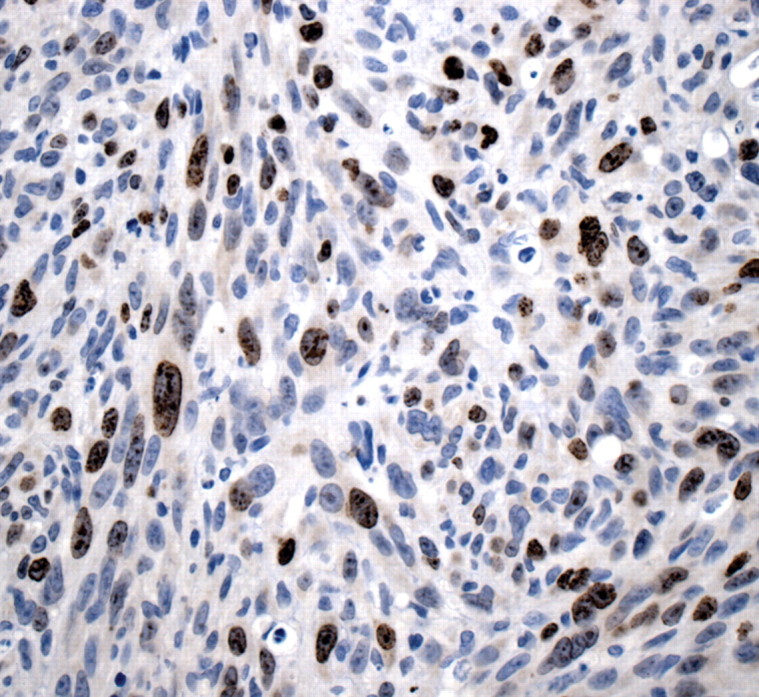
Figures
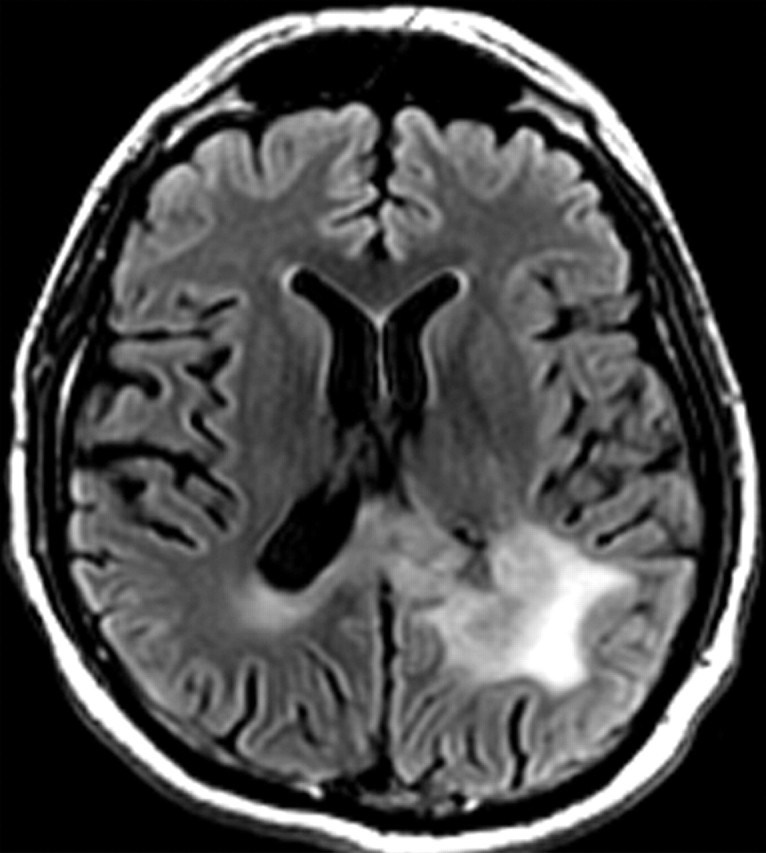
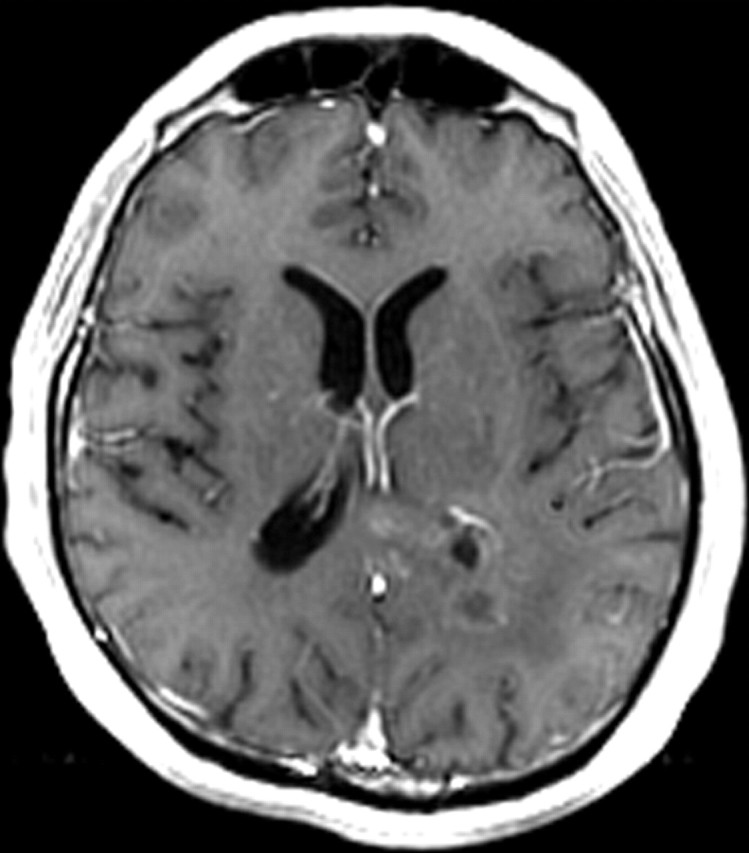
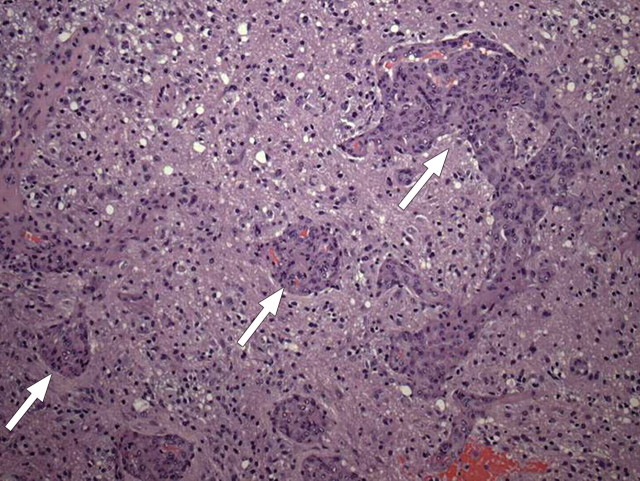
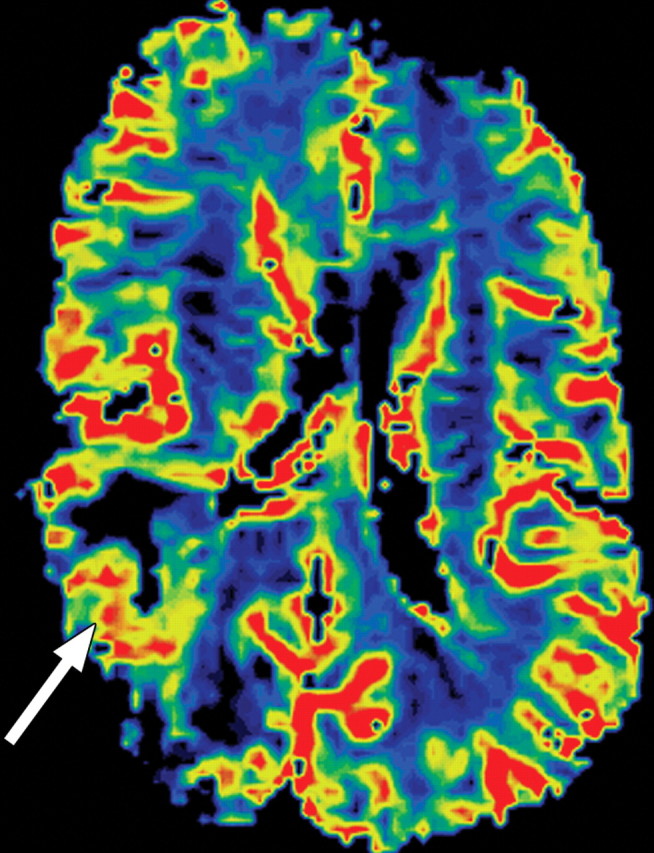
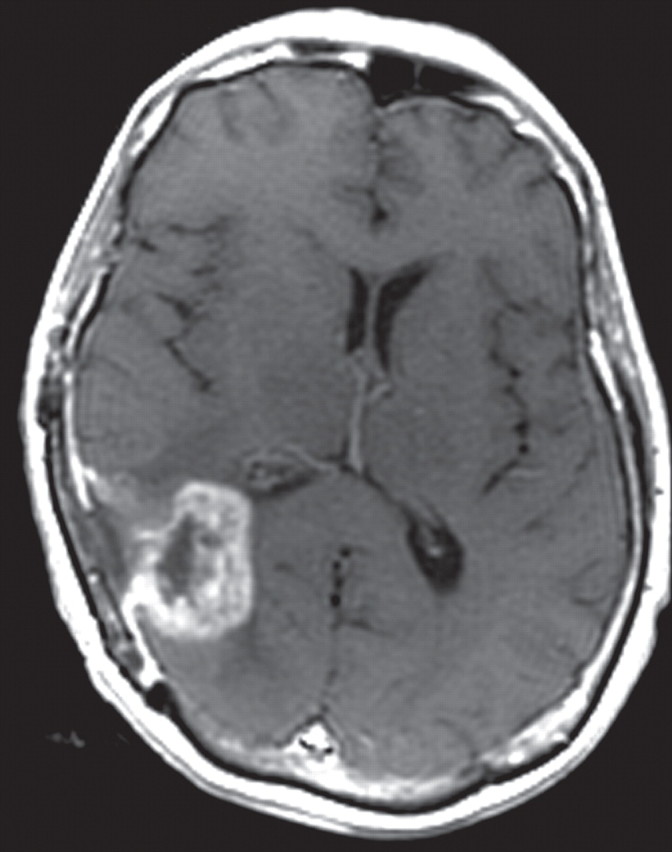
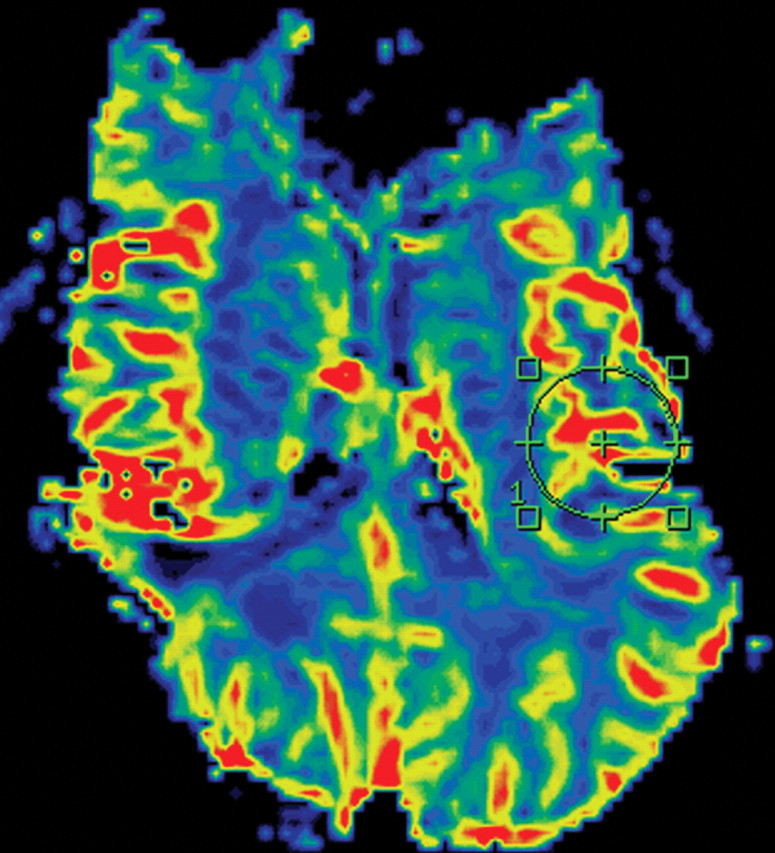
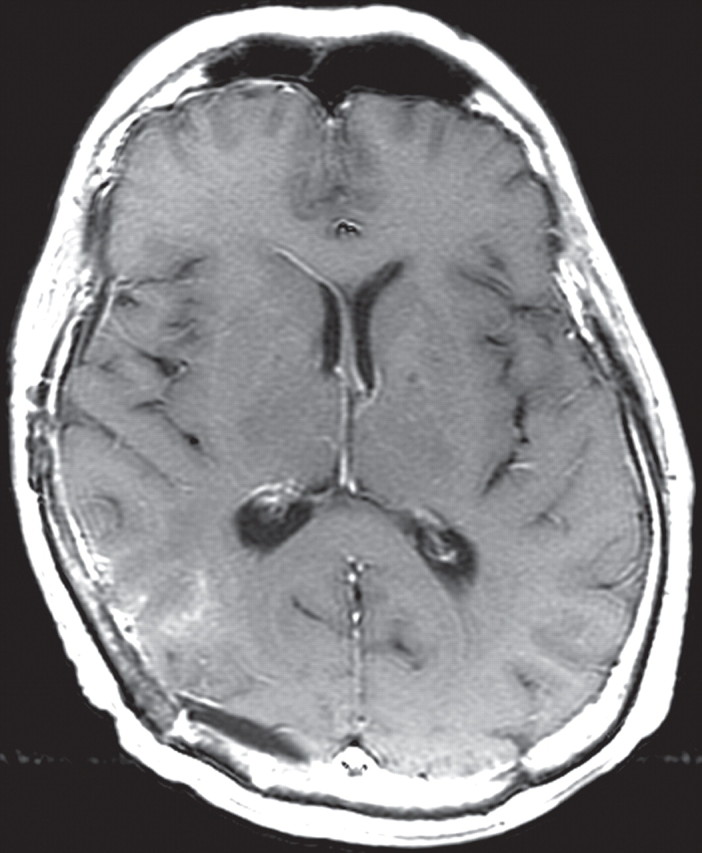
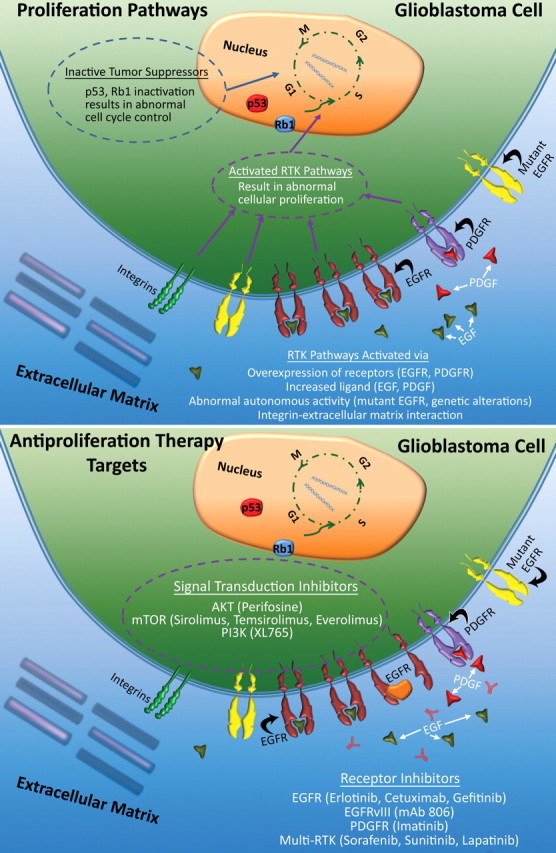
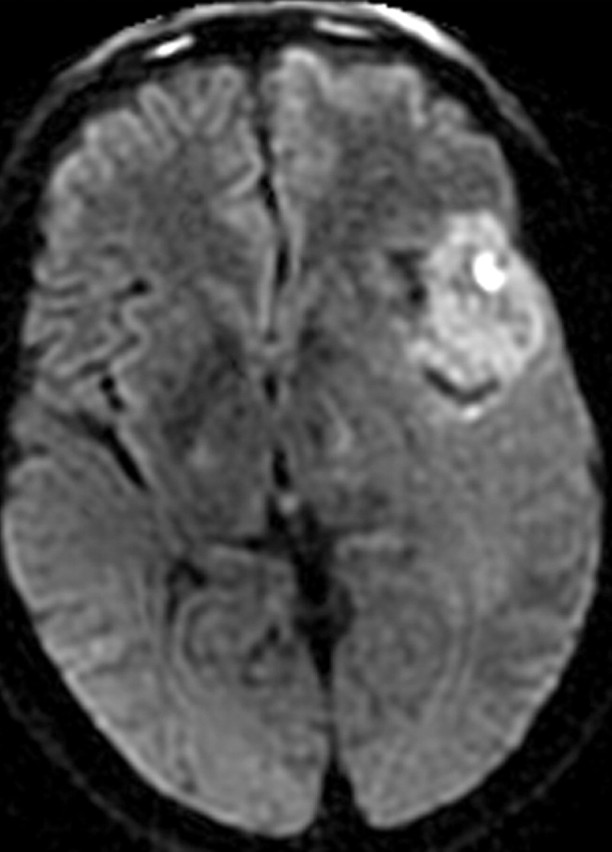
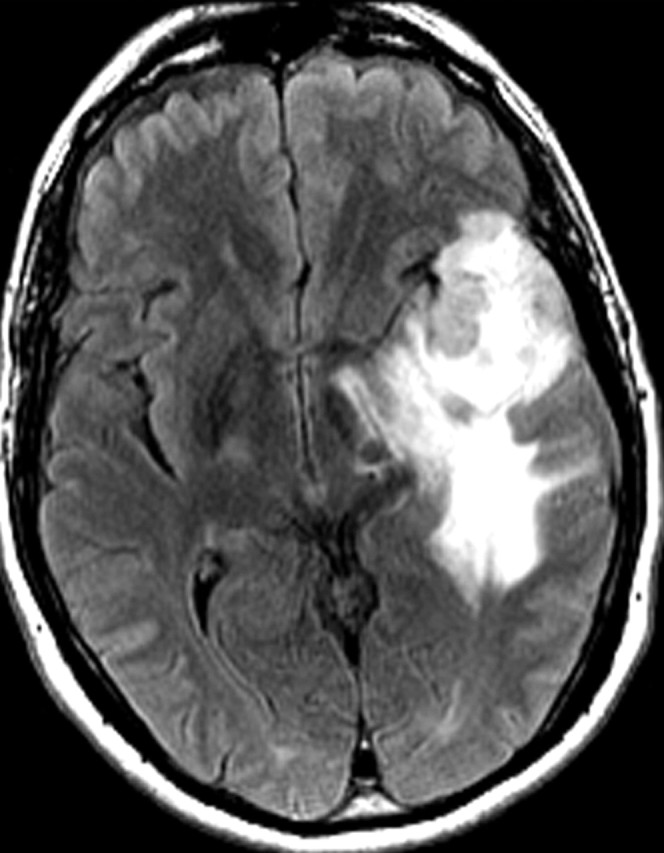
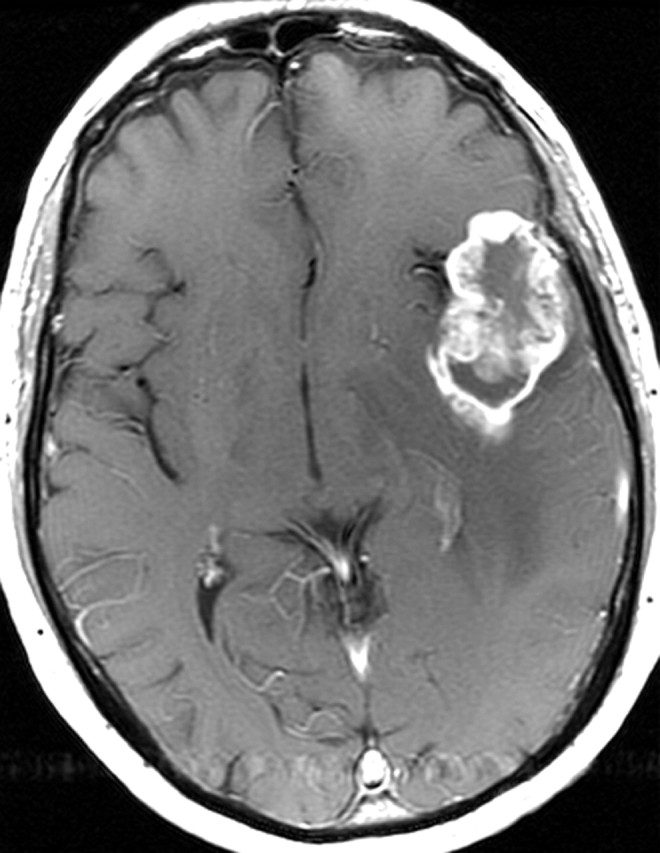
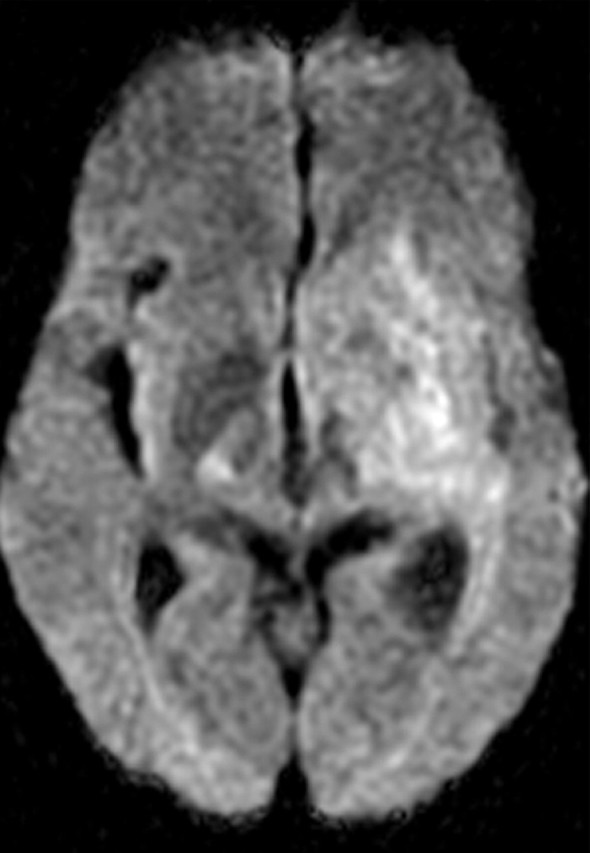
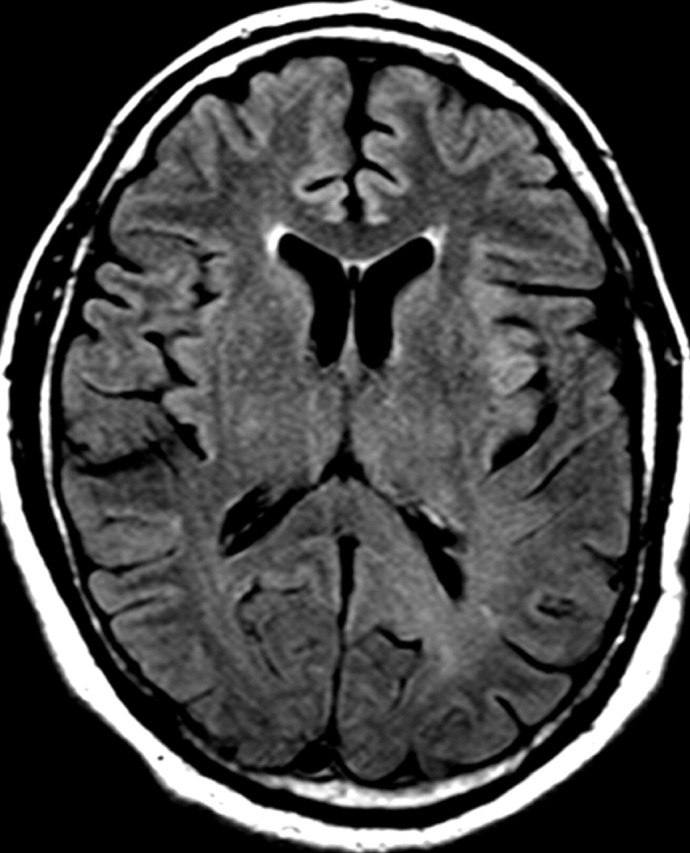
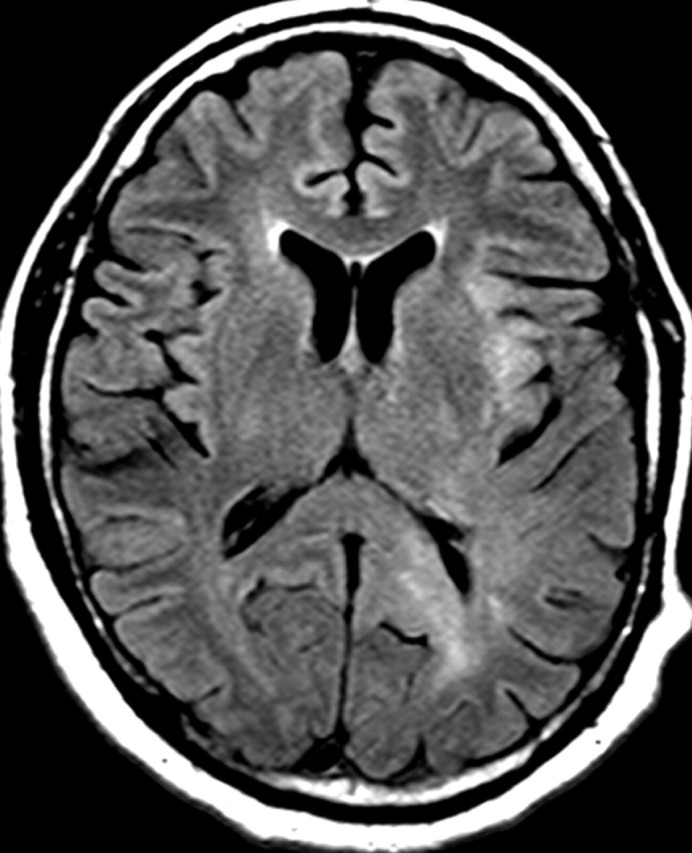
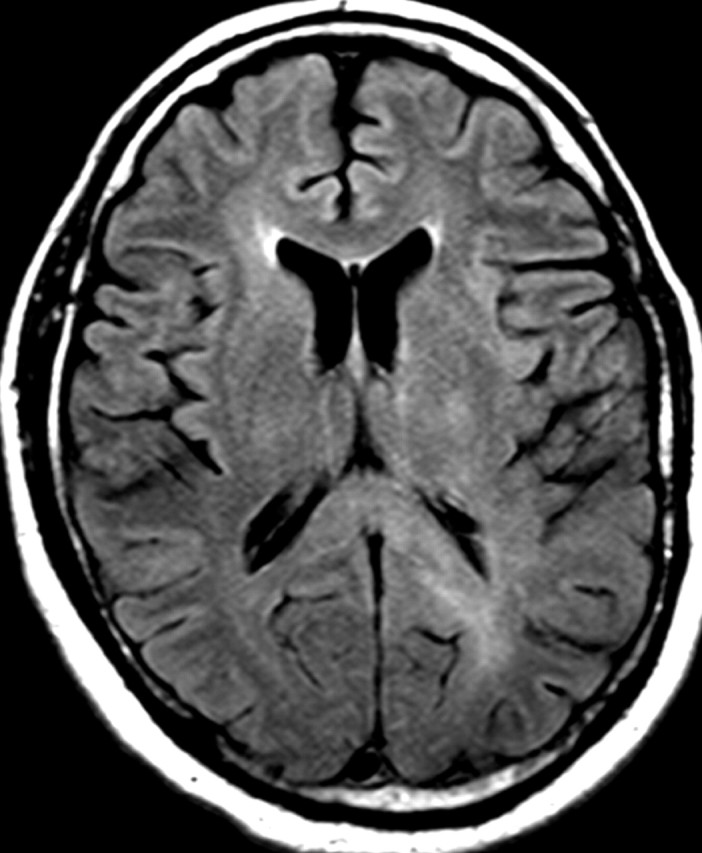
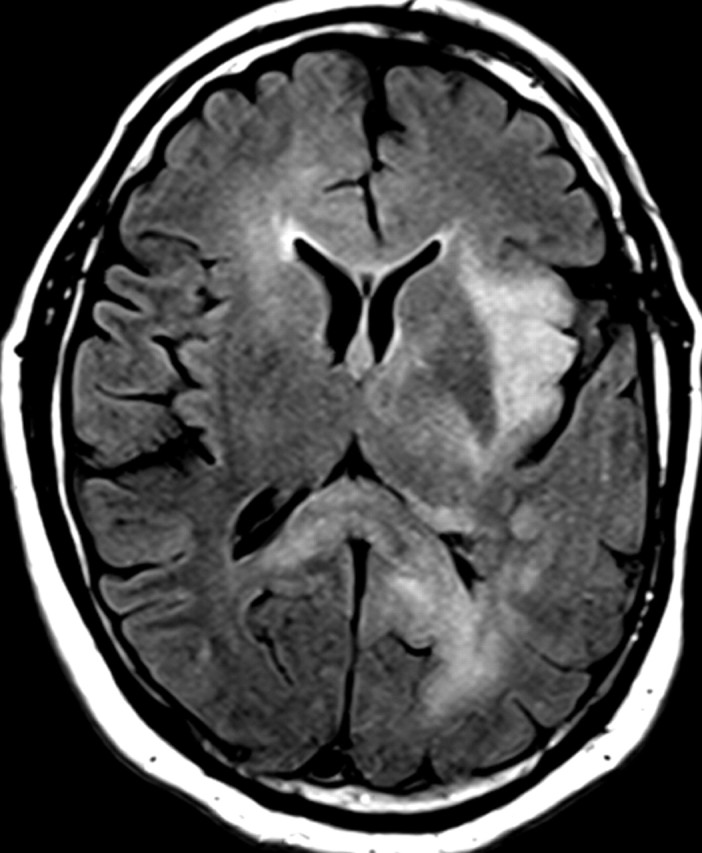
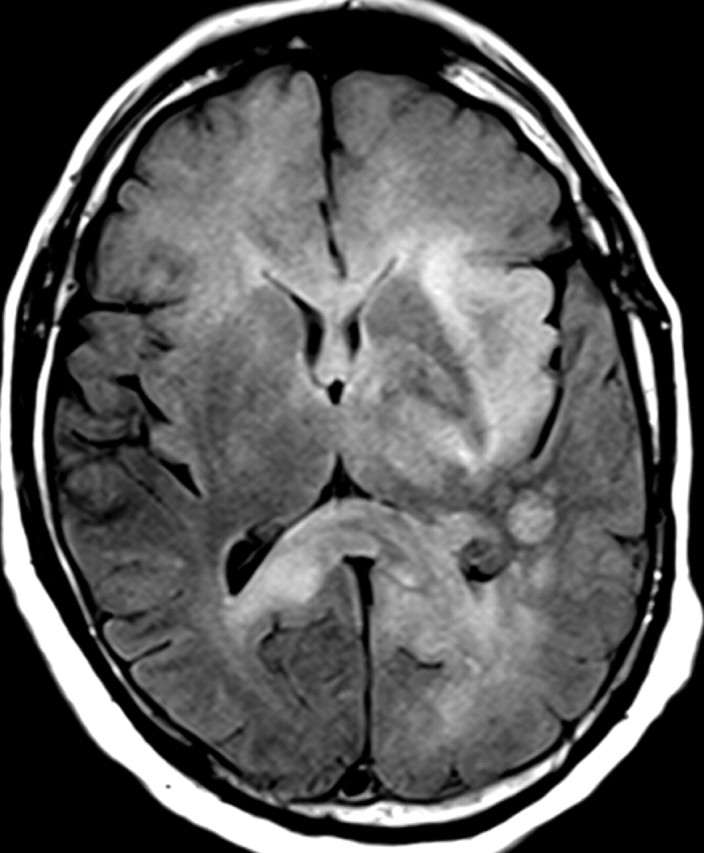
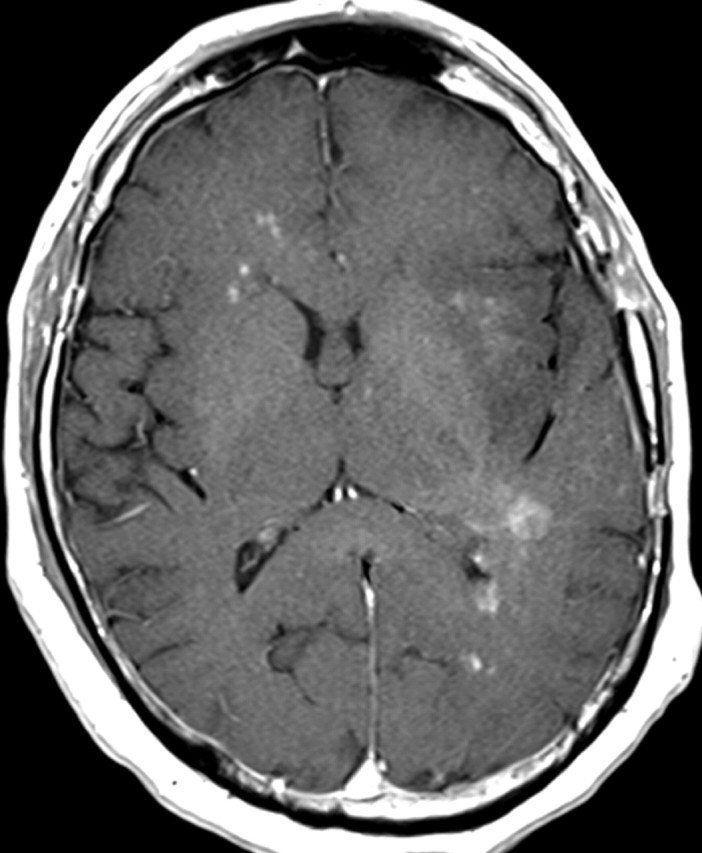
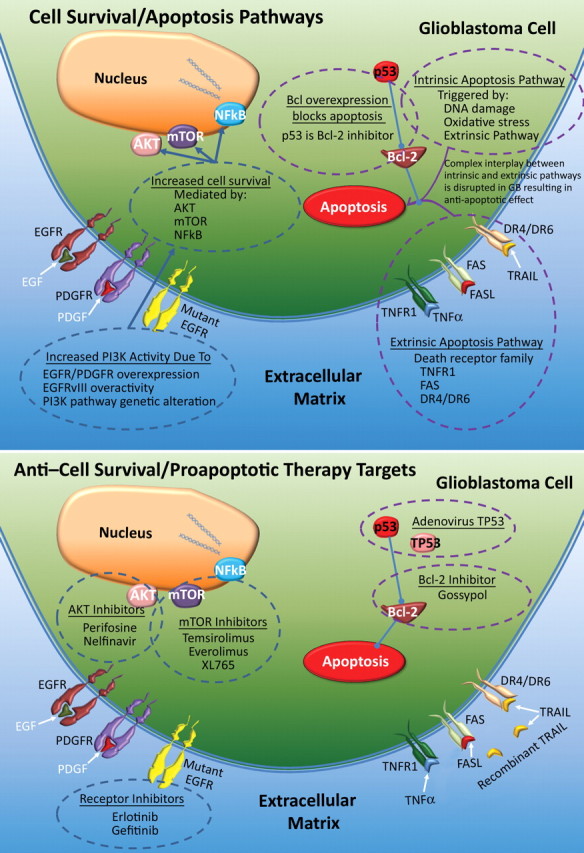
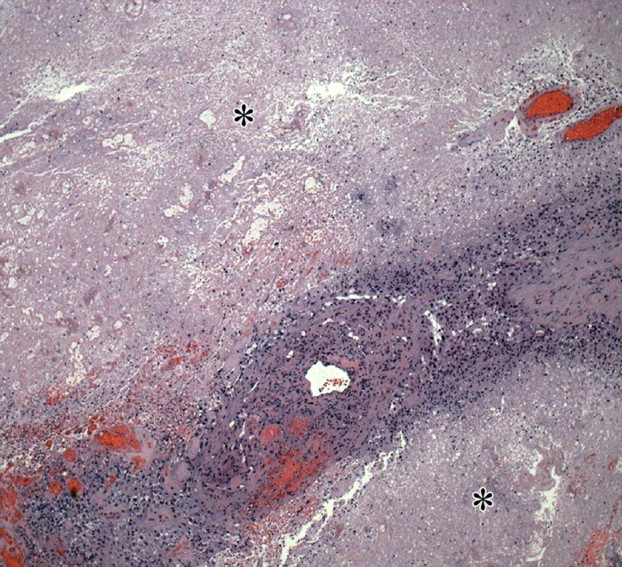
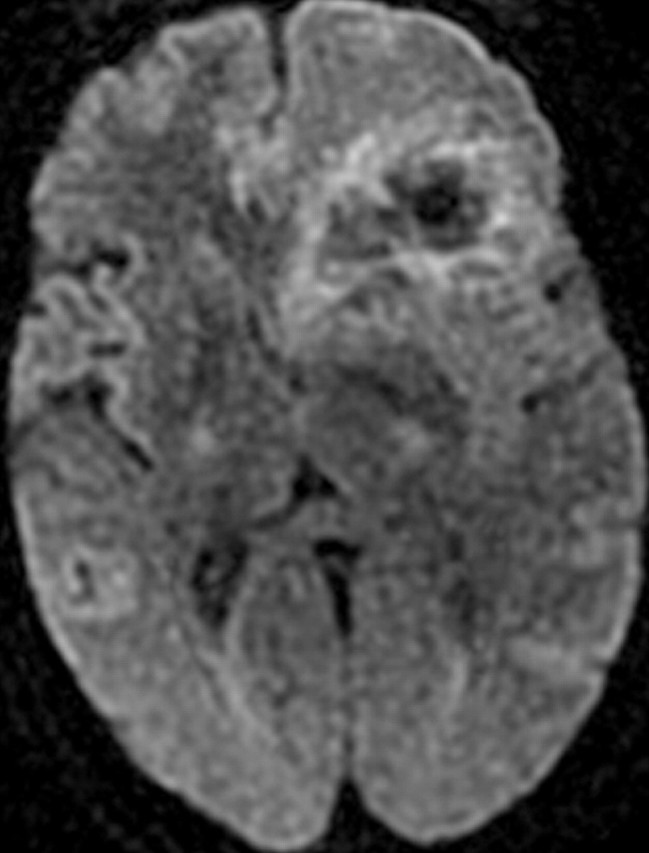
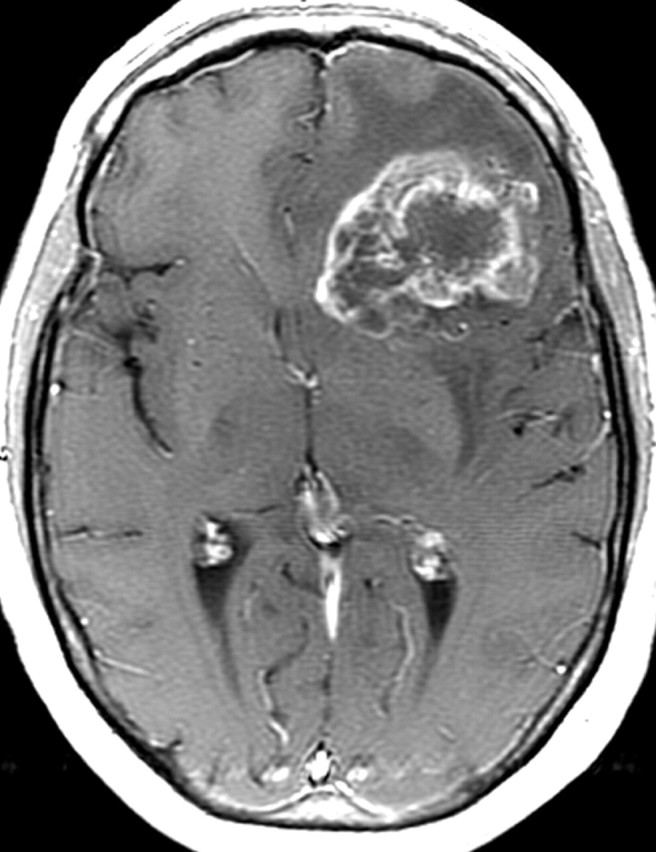
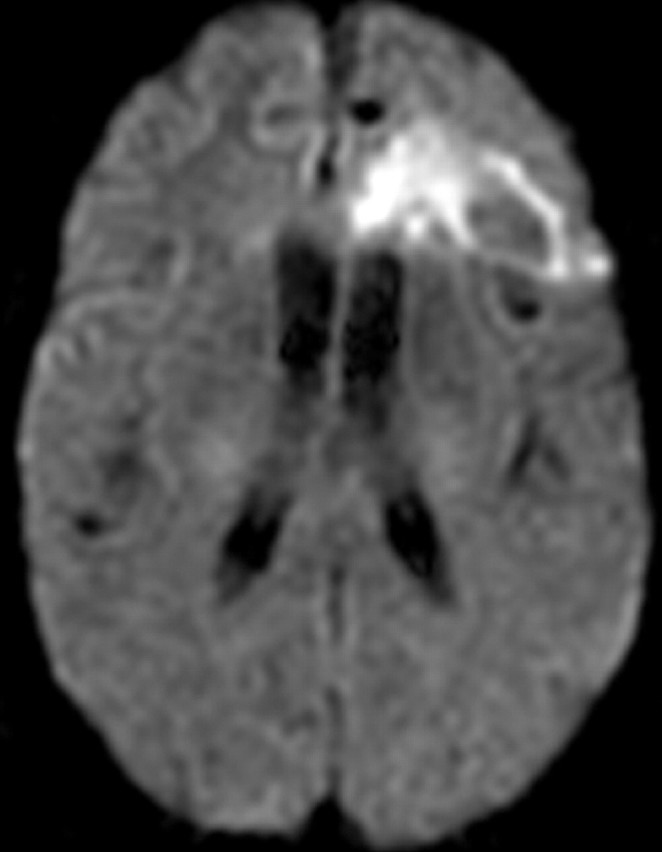
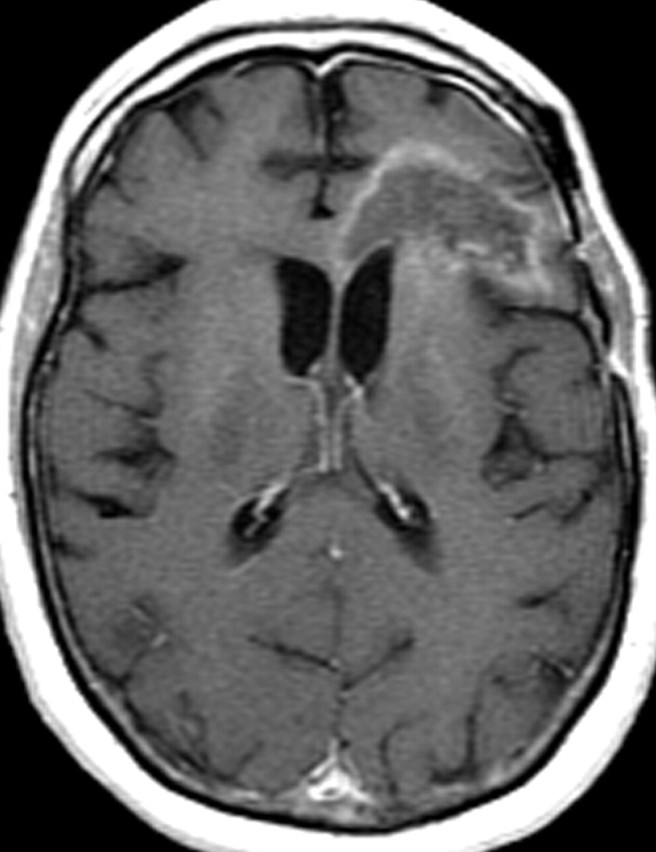
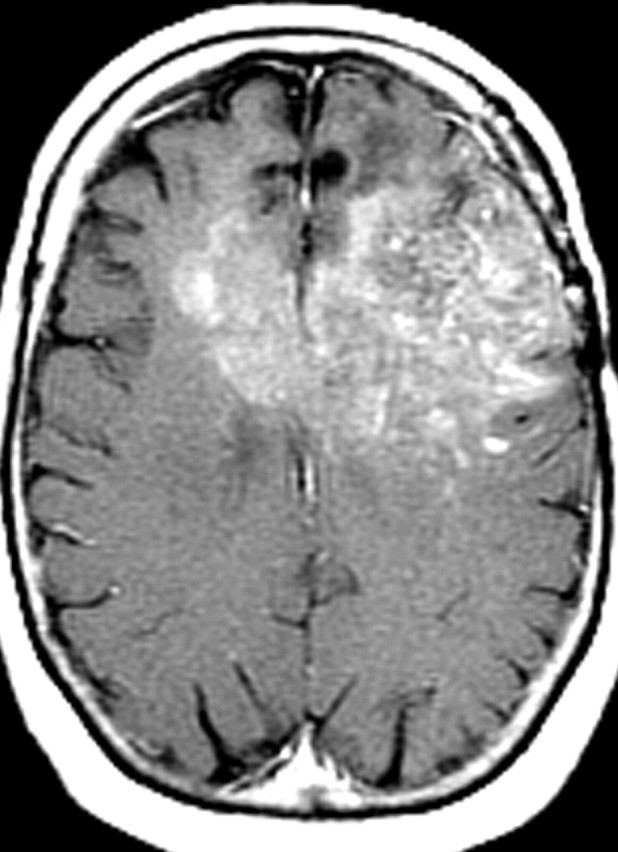
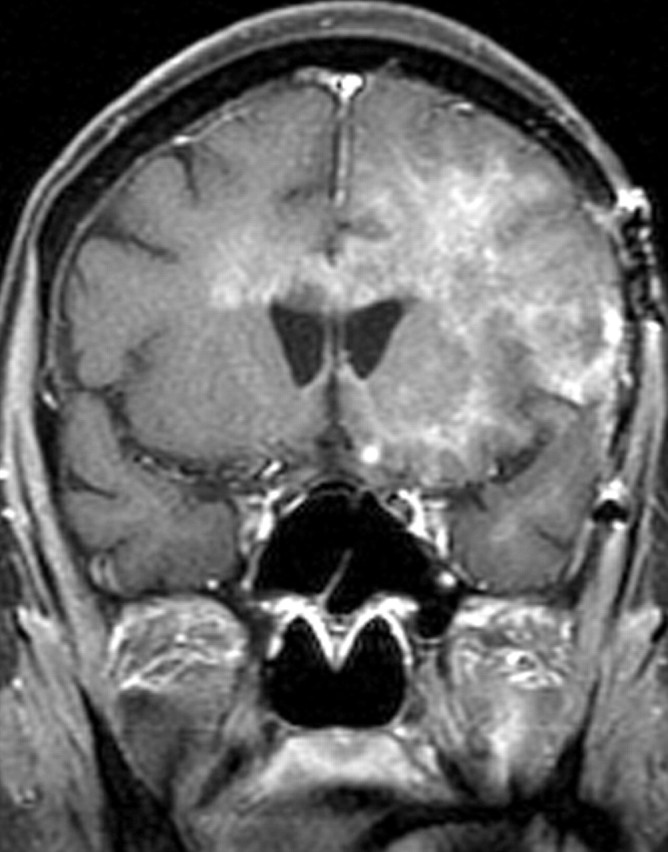
References
-
- Furnari FB, Fenton T, Bachoo RM, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev 2007;21(21):2683–2710. - PubMed
-
- Rao SK, Edwards J, Joshi AD, Siu IM, Riggins GJ. A survey of glioblastoma genomic amplifications and deletions. J Neurooncol 2010;96(2):169–179. - PubMed
-
- Jouanneau E. Angiogenesis and gliomas: current issues and development of surrogate markers. Neurosurgery 2008;62(1):31–50; discussion 50–52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
