

A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity
- PMID: 21998323
- PMCID: PMC3908774
- DOI: 10.1161/CIRCRESAHA.111.255695
A novel preclinical strategy for identifying cardiotoxic kinase inhibitors and mechanisms of cardiotoxicity
Abstract
Rationale: Despite intense interest in strategies to predict which kinase inhibitor (KI) cancer therapeutics may be associated with cardiotoxicity, current approaches are inadequate. Sorafenib is a KI of concern because it inhibits growth factor receptors and Raf-1/B-Raf, kinases that are upstream of extracellular signal-regulated kinases (ERKs) and signal cardiomyocyte survival in the setting of stress.
Objectives: To explore the potential use of zebrafish as a preclinical model to predict cardiotoxicity and to determine whether sorafenib has associated cardiotoxicity, and, if so, define the mechanisms.
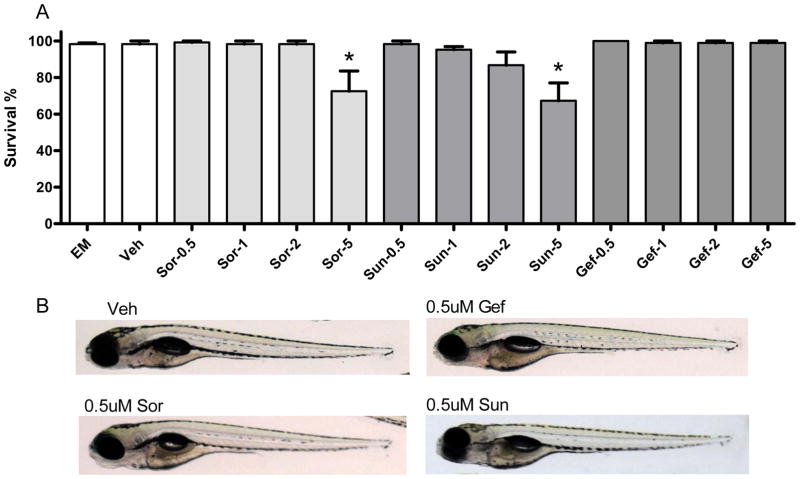
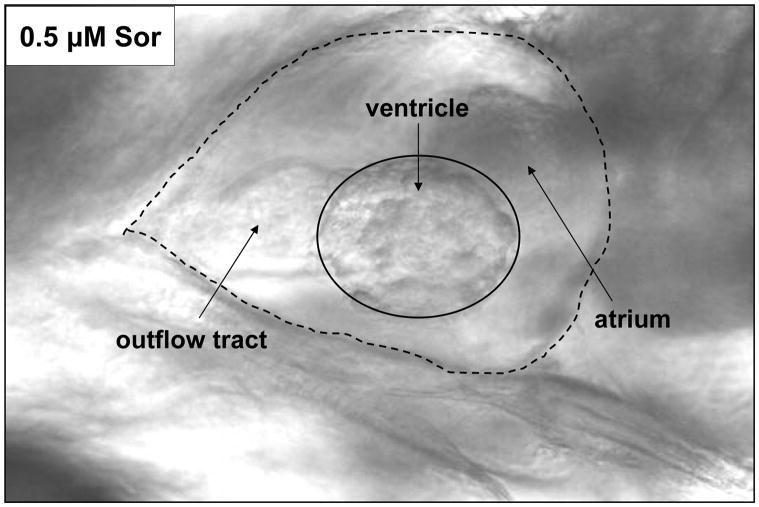
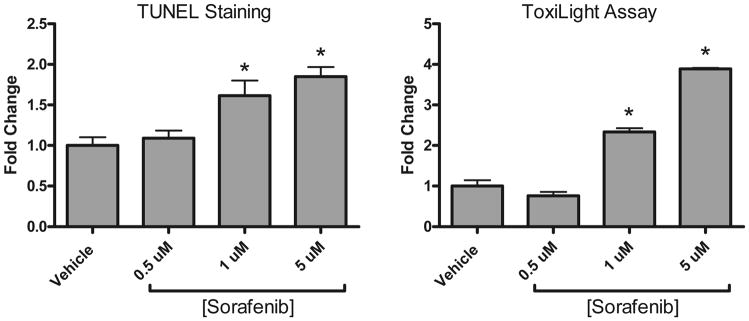
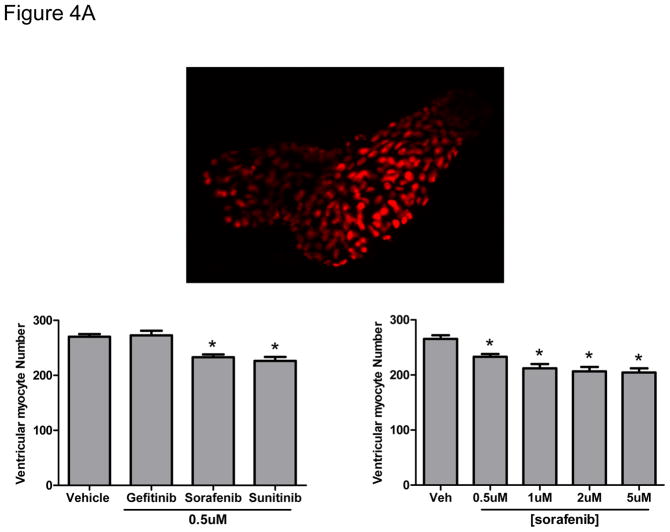
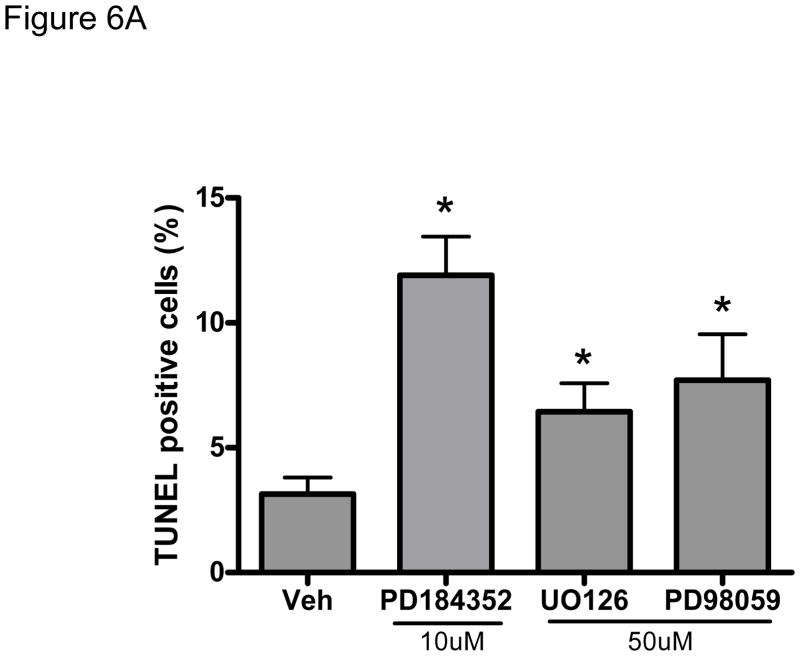
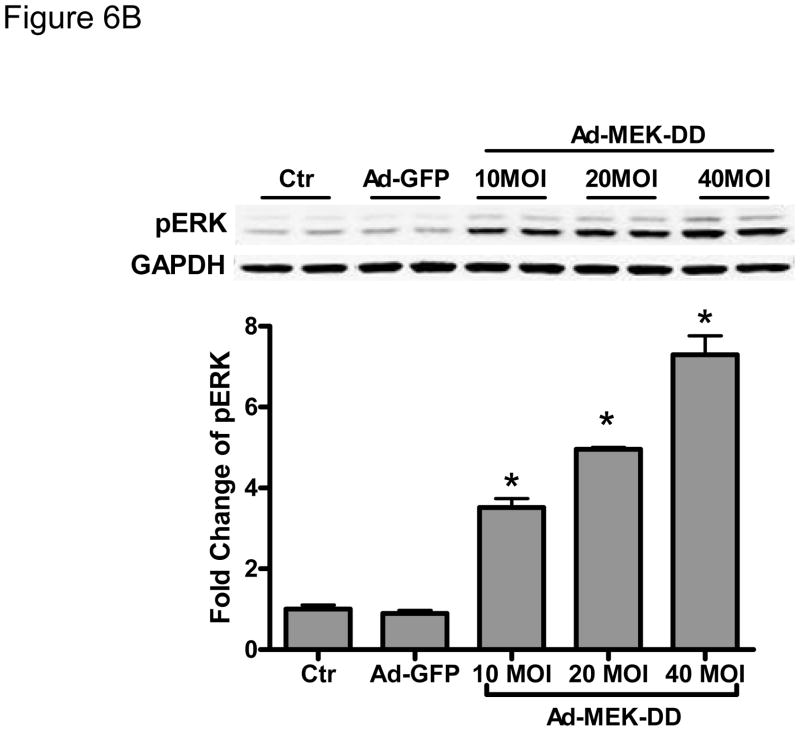
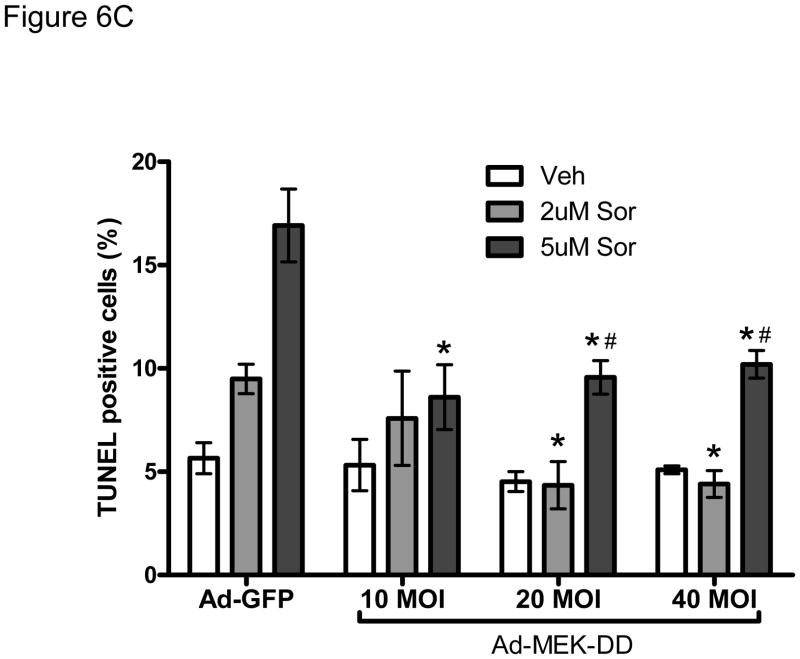
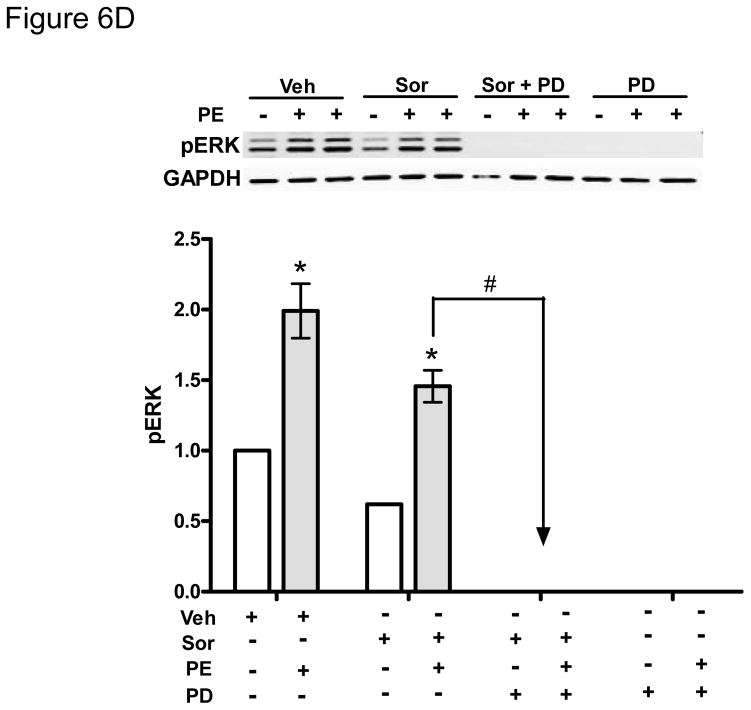
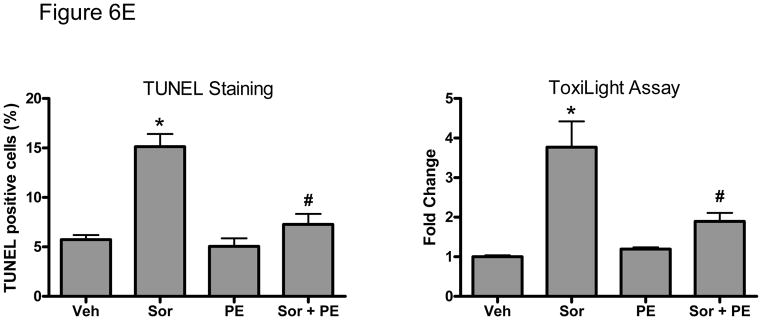
Methods and results: We find that the zebrafish model is readily able to discriminate a KI with little or no cardiotoxicity (gefitinib) from one with demonstrated cardiotoxicity (sunitinib). Sorafenib, like sunitinib, leads to cardiomyocyte apoptosis, a reduction in total myocyte number per heart, contractile dysfunction, and ventricular dilatation in zebrafish. In cultured rat cardiomyocytes, sorafenib induces cell death. This can be rescued by adenovirus-mediated gene transfer of constitutively active MEK1, which restores ERK activity even in the presence of sorafenib. Whereas growth factor-induced activation of ERKs requires Raf, α-adrenergic agonist-induced activation of ERKs does not require it. Consequently, activation of α-adrenergic signaling markedly decreases sorafenib-induced cell death. Consistent with these in vitro data, inhibition of α-adrenergic signaling with the receptor antagonist prazosin worsens sorafenib-induced cardiomyopathy in zebrafish.
Conclusions: Zebrafish may be a valuable preclinical tool to predict cardiotoxicity. The α-adrenergic signaling pathway is an important modulator of sorafenib cardiotoxicity in vitro and in vivo and appears to act through a here-to-fore unrecognized signaling pathway downstream of α-adrenergic activation that bypasses Raf to activate ERKs.
Figures
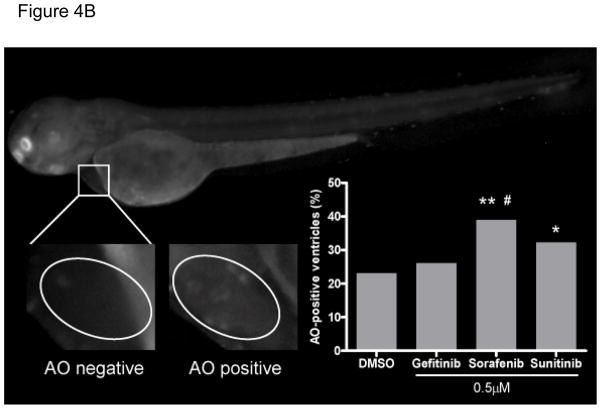
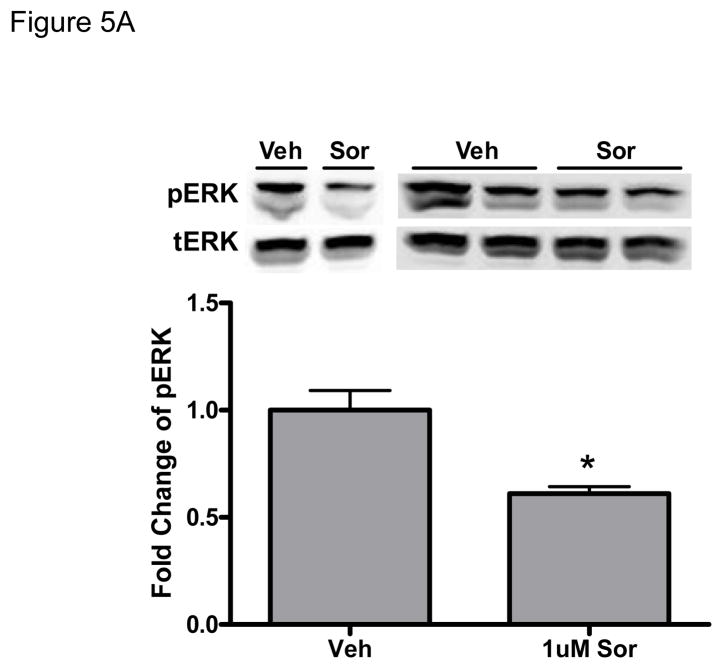
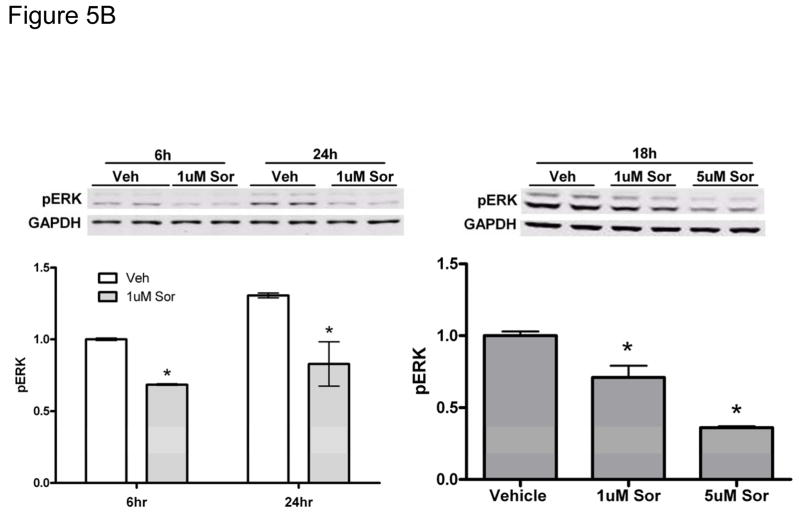
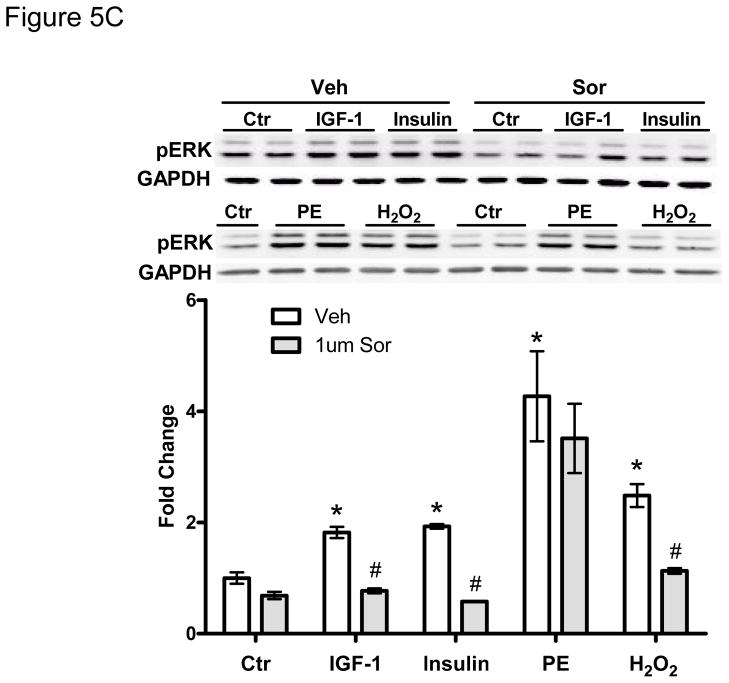
References
-
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. - PubMed
-
- Chu TF, Rupnick MA, Kerkela R, Dallabrida SM, Zurakowski D, Nguyen L, Woulfe K, Pravda E, Cassiola F, Desai J, George S, Morgan JA, Harris DM, Ismail NS, Chen JH, Schoen FJ, Van den Abbeele AD, Demetri GD, Force T, Chen MH. Cardiotoxicity associated with tyrosine kinase inhibitor sunitinib. Lancet. 2007 Dec 15;370(9604):2011–2019. - PMC - PubMed
-
- Chico TJ, Ingham PW, Crossman DC. Modeling cardiovascular disease in the zebrafish. Trends Cardiovasc Med. 2008 May;18(4):150–155. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
