

Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis
- PMID: 22000011
- PMCID: PMC3202669
- DOI: 10.1016/j.cell.2011.08.046
Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis
Abstract
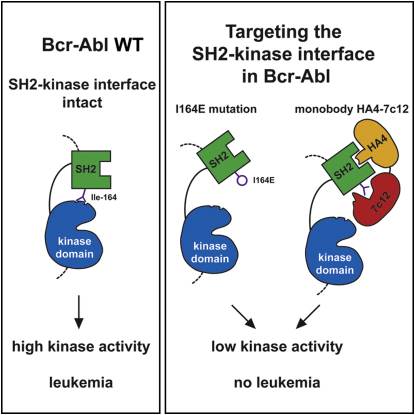
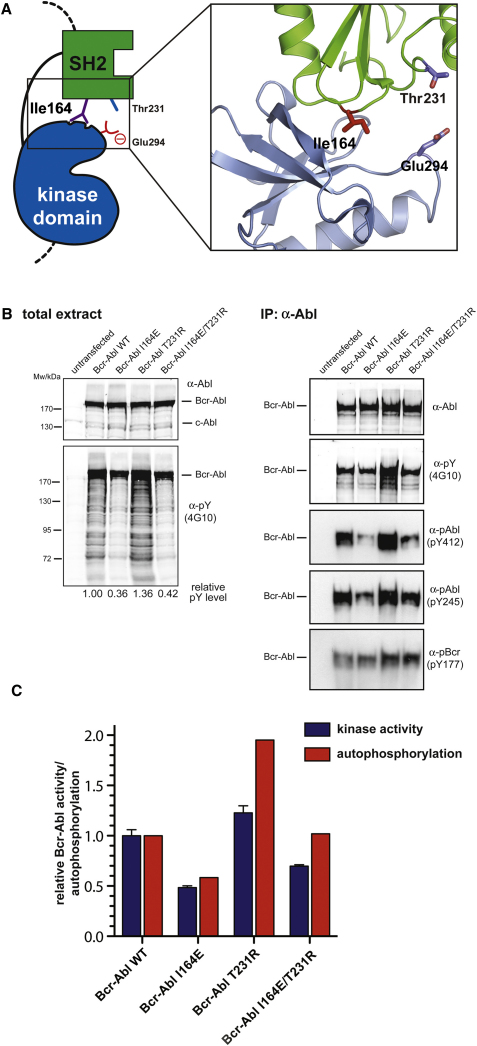
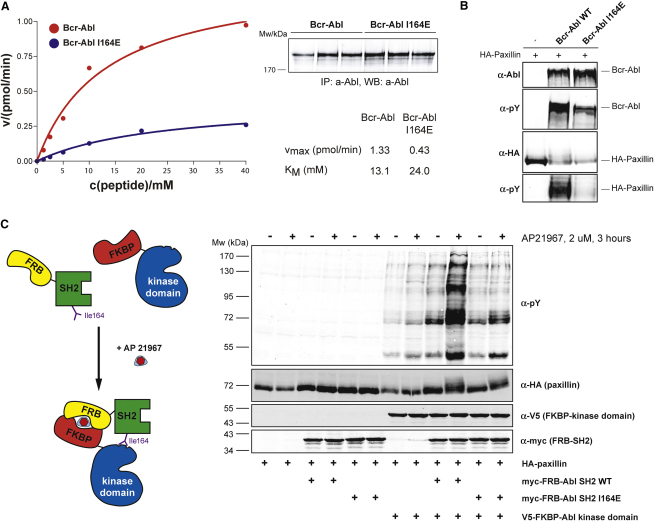
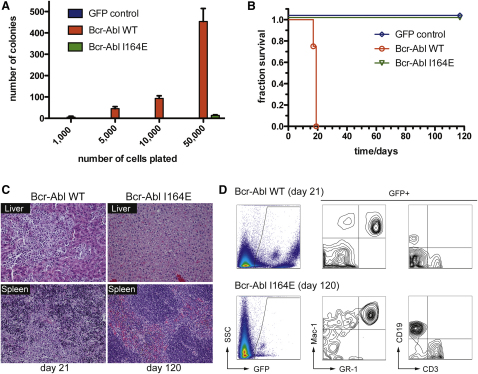
Chronic myelogenous leukemia (CML) is caused by the constitutively active tyrosine kinase Bcr-Abl and treated with the tyrosine kinase inhibitor (TKI) imatinib. However, emerging TKI resistance prevents complete cure. Therefore, alternative strategies targeting regulatory modules of Bcr-Abl in addition to the kinase active site are strongly desirable. Here, we show that an intramolecular interaction between the SH2 and kinase domains in Bcr-Abl is both necessary and sufficient for high catalytic activity of the enzyme. Disruption of this interface led to inhibition of downstream events critical for CML signaling and, importantly, completely abolished leukemia formation in mice. Furthermore, disruption of the SH2-kinase interface increased sensitivity of imatinib-resistant Bcr-Abl mutants to TKI inhibition. An engineered Abl SH2-binding fibronectin type III monobody inhibited Bcr-Abl kinase activity both in vitro and in primary CML cells, where it induced apoptosis. This work validates the SH2-kinase interface as an allosteric target for therapeutic intervention.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
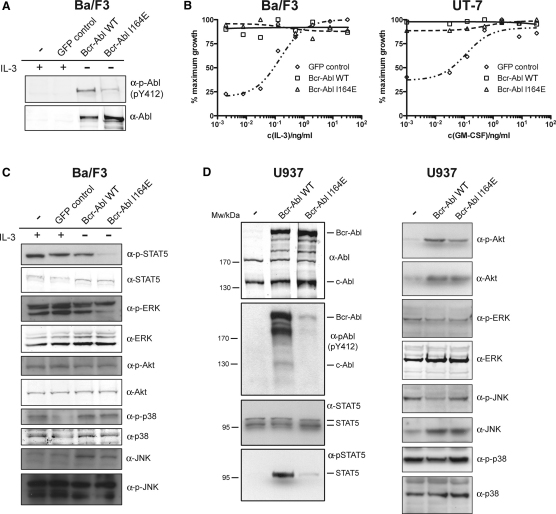
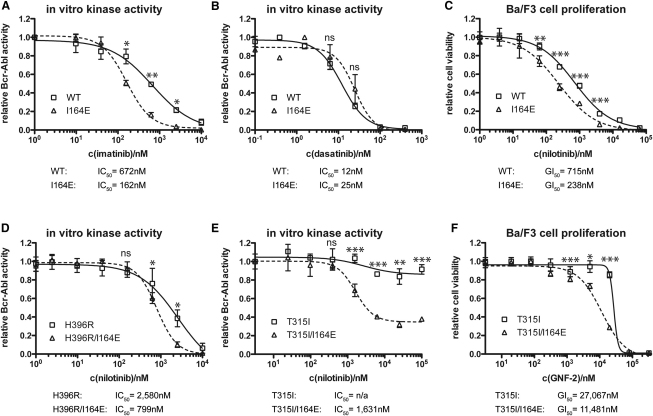
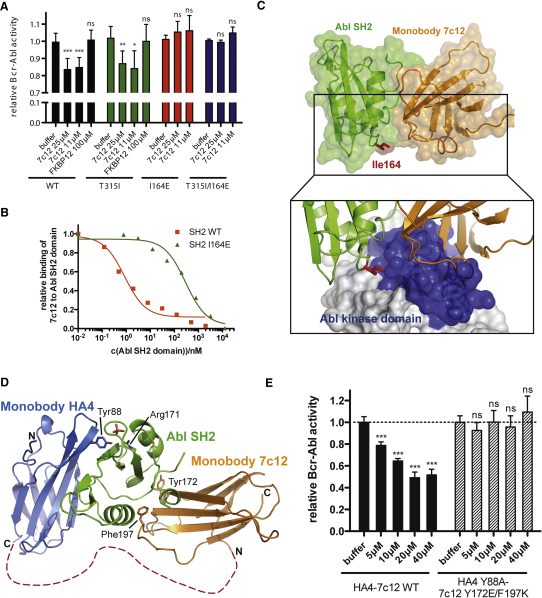
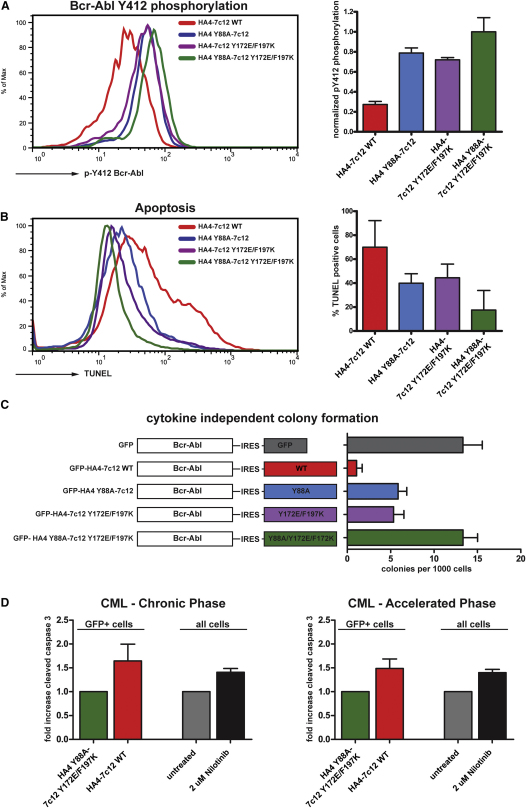
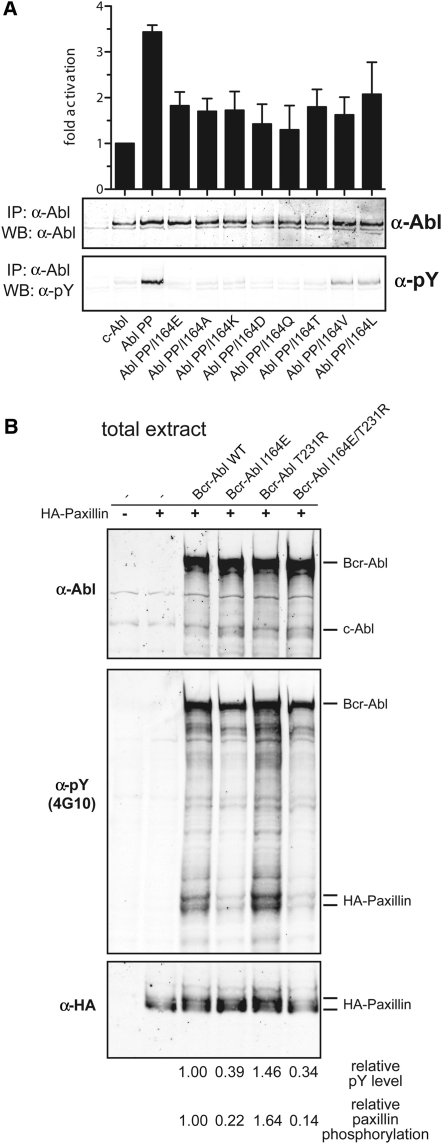
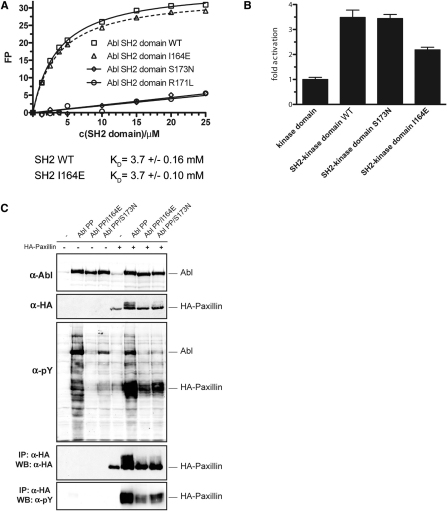
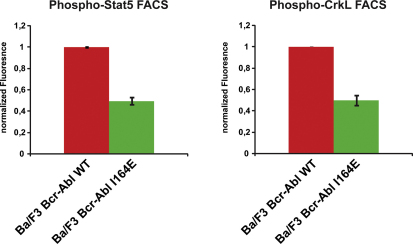
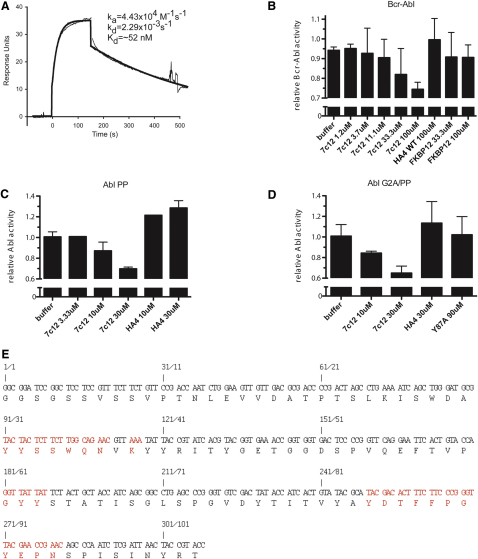
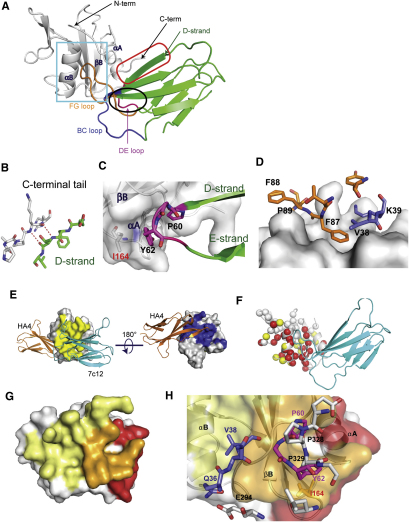
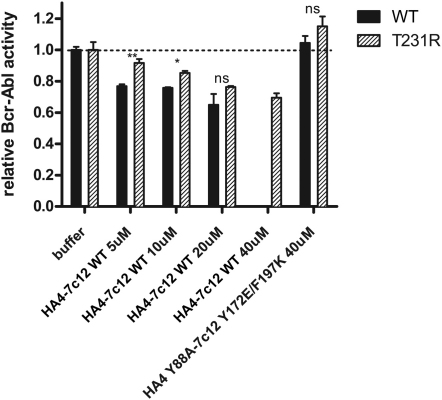
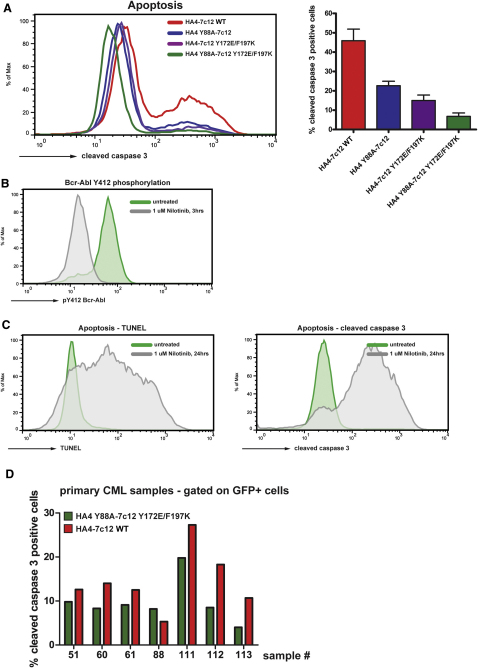
Comment in
-
Therapeutics. Another tool in the BCR-ABL kit?Nat Rev Cancer. 2011 Nov 10;11(12):832-3. doi: 10.1038/nrc3173. Nat Rev Cancer. 2011. PMID: 22071978 No abstract available.
-
BCR-ABL1 kinase: hunting an elusive target with new weapons.Chem Biol. 2011 Nov 23;18(11):1352-3. doi: 10.1016/j.chembiol.2011.11.001. Chem Biol. 2011. PMID: 22118668 Free PMC article.
References
-
- Daley G.Q., Van Etten R.A., Baltimore D. Induction of chronic myelogenous leukemia in mice by the P210bcr/abl gene of the Philadelphia chromosome. Science. 1990;247:824–830. - PubMed
-
- De Keersmaecker K., Versele M., Cools J., Superti-Furga G., Hantschel O. Intrinsic differences between the catalytic properties of the oncogenic NUP214-ABL1 and BCR-ABL1 fusion protein kinases. Leukemia. 2008;22:2208–2216. - PubMed
-
- Deininger M., Buchdunger E., Druker B.J. The development of imatinib as a therapeutic agent for chronic myeloid leukemia. Blood. 2005;105:2640–2653. - PubMed
Supplemental References
-
- Collaborative Computational Project, Number 4. (1994). The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763. - PubMed
-
- Barilá, D., and Superti-Furga, G. (1998). An intramolecular SH3-domain interaction regulates c-Abl activity. Nat. Genet. 18, 280–282. - PubMed
-
- Davis, I.W., Leaver-Fay, A., Chen, V.B., Block, J.N., Kapral, G.J., Wang, X., Murray, L.W., Arendall, W.B., 3rd, Snoeyink, J., Richardson, J.S., and Richardson, D.C. (2007). MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35 (Web Server issue), W375–W383. - PMC - PubMed
-
- DeLano, W.L. (2002). The PyMOL Molecular Graphics System. http://pymol.sourceforge.net.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
