

Systematic analysis of protein pools, isoforms, and modifications affecting turnover and subcellular localization
- PMID: 22002106
- PMCID: PMC3316725
- DOI: 10.1074/mcp.M111.013680
Systematic analysis of protein pools, isoforms, and modifications affecting turnover and subcellular localization
Abstract
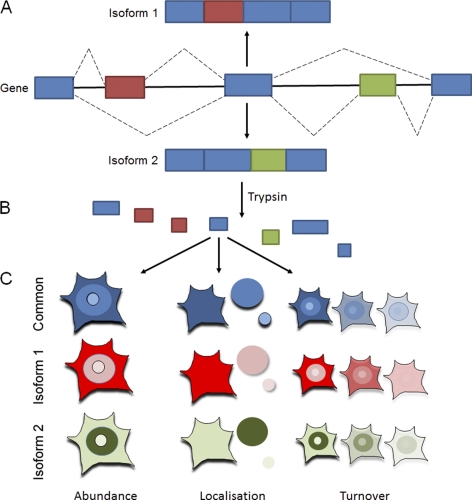
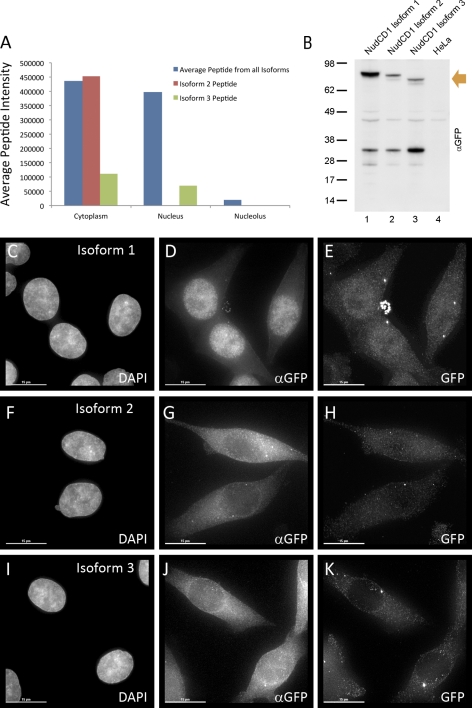
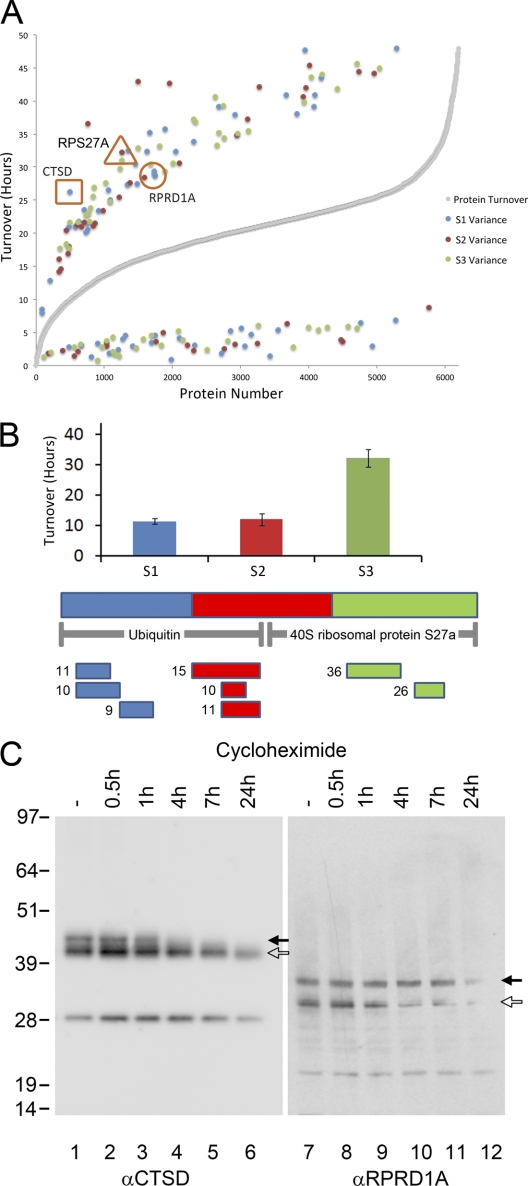
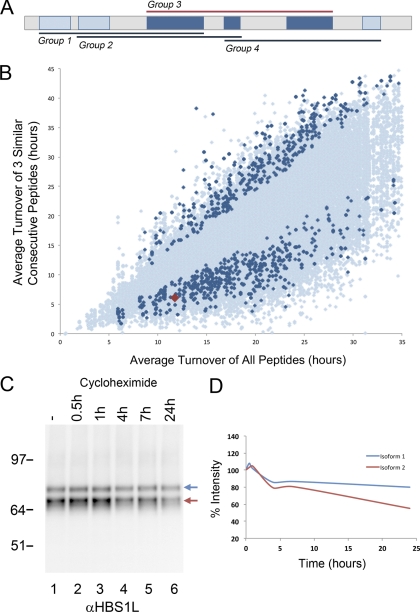
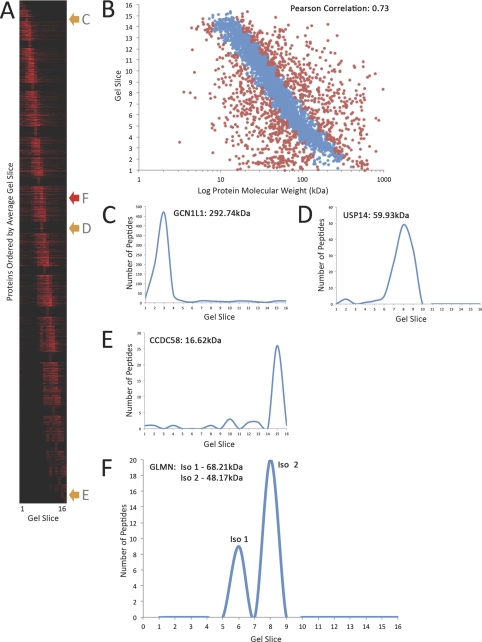
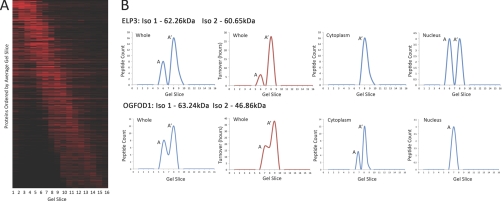
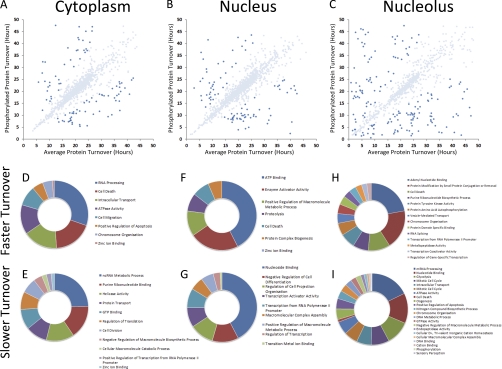
In higher eukaryotes many genes encode protein isoforms whose properties and biological roles are often poorly characterized. Here we describe systematic approaches for detection of either distinct isoforms, or separate pools of the same isoform, with differential biological properties. Using information from ion intensities we have estimated protein abundance levels and using rates of change in stable isotope labeling with amino acids in cell culture isotope ratios we measured turnover rates and subcellular distribution for the HeLa cell proteome. Protein isoforms were detected using three data analysis strategies that evaluate differences between stable isotope labeling with amino acids in cell culture isotope ratios for specific groups of peptides within the total set of peptides assigned to a protein. The candidate approach compares stable isotope labeling with amino acids in cell culture isotope ratios for predicted isoform-specific peptides, with ratio values for peptides shared by all the isoforms. The rule of thirds approach compares the mean isotope ratio values for all peptides in each of three equal segments along the linear length of the protein, assessing differences between segment values. The three in a row approach compares mean isotope ratio values for each sequential group of three adjacent peptides, assessing differences with the mean value for all peptides assigned to the protein. Protein isoforms were also detected and their properties evaluated by fractionating cell extracts on one-dimensional SDS-PAGE prior to trypsin digestion and MS analysis and independently evaluating isotope ratio values for the same peptides isolated from different gel slices. The effect of protein phosphorylation on turnover rates was analyzed by comparing mean turnover values calculated for all peptides assigned to a protein, either including, or excluding, values for cognate phosphopeptides. Collectively, these experimental and analytical approaches provide a framework for expanding the functional annotation of the genome.
Figures
References
-
- Corthals G. L., Wasinger V. C., Hochstrasser D. F., Sanchez J. C. (2000) The dynamic range of protein expression: a challenge for proteomic research. Electrophoresis 21, 1104–1115 - PubMed
-
- Hinkson I. V., Elias J. E. (2011) The dynamic state of protein turnover: It's about time. Trends Cell. Biol. 21, 293–303 - PubMed
-
- Nesvizhskii A. I., Aebersold R. (2005) Interpretation of Shotgun Proteomic Data. Mol. Cell. Proteomics 4, 1419–1440 - PubMed
-
- Rappsilber J., Mann M. (2002) What does it mean to identify a protein in proteomics? Trends Biochem. Sci. 27, 74–78 - PubMed
-
- Godovac-Zimmermann J., Kleiner O., Brown L. R., Drukier A. K. (2005) Perspectives in spicing up proteomics with splicing. Proteomics 5, 699–709 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
