

Gcn5 loss-of-function accelerates cerebellar and retinal degeneration in a SCA7 mouse model
- PMID: 22002997
- PMCID: PMC3276287
- DOI: 10.1093/hmg/ddr474
Gcn5 loss-of-function accelerates cerebellar and retinal degeneration in a SCA7 mouse model
Abstract
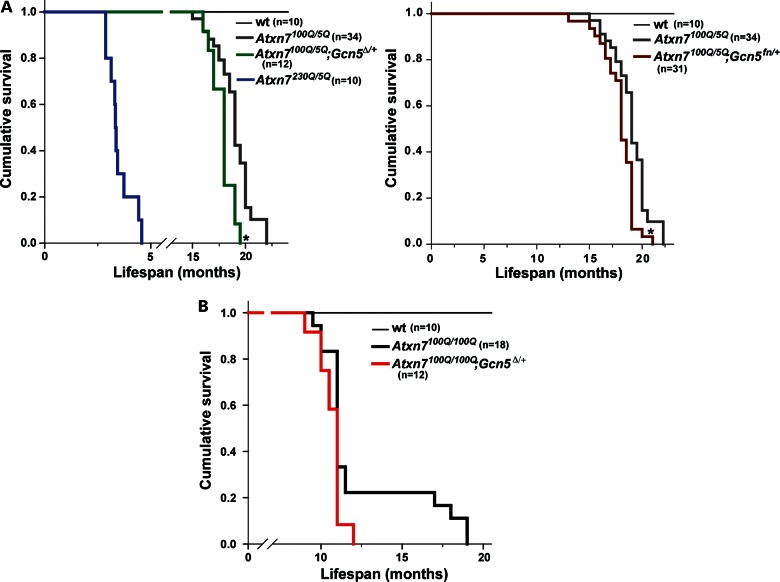
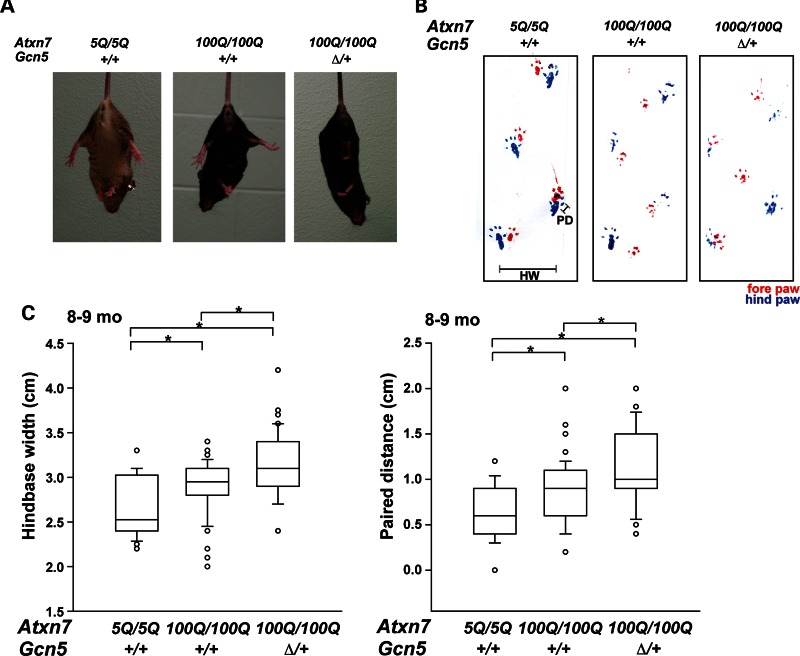
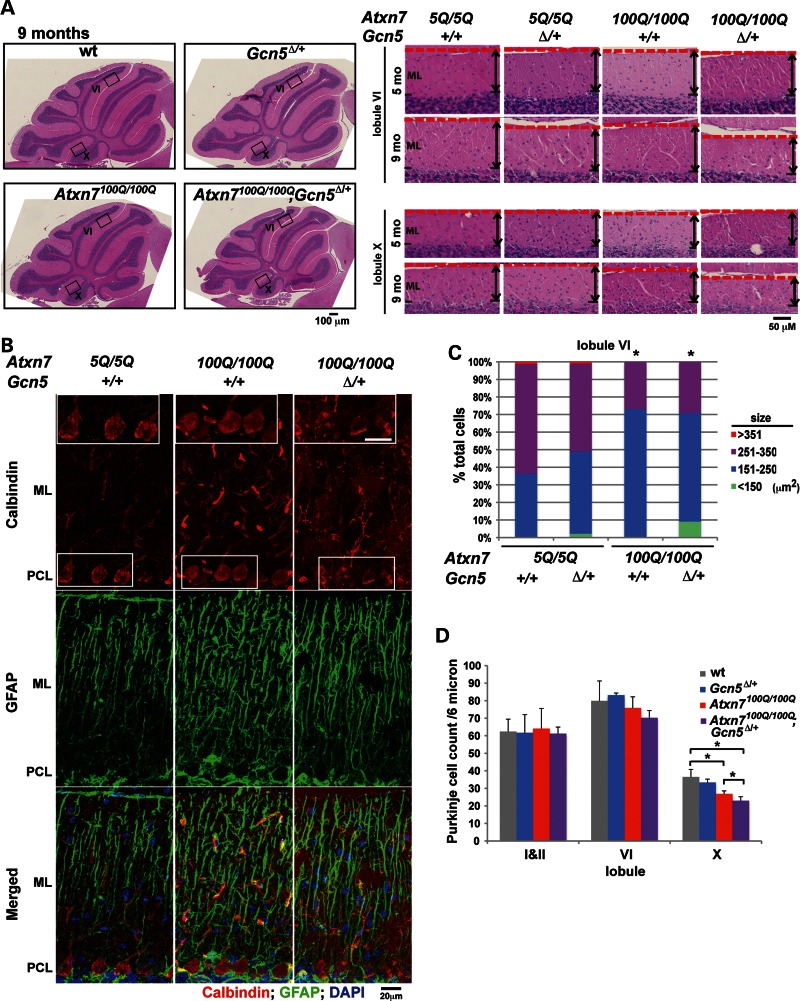
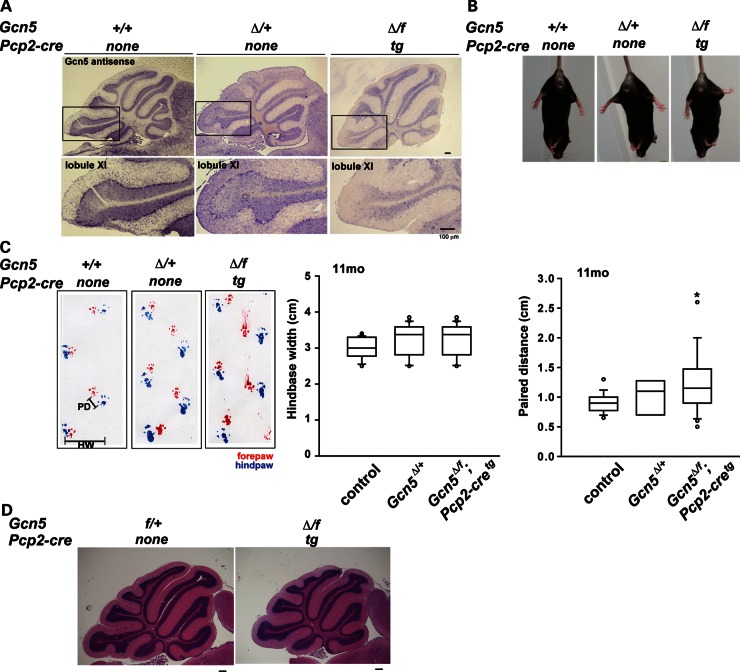
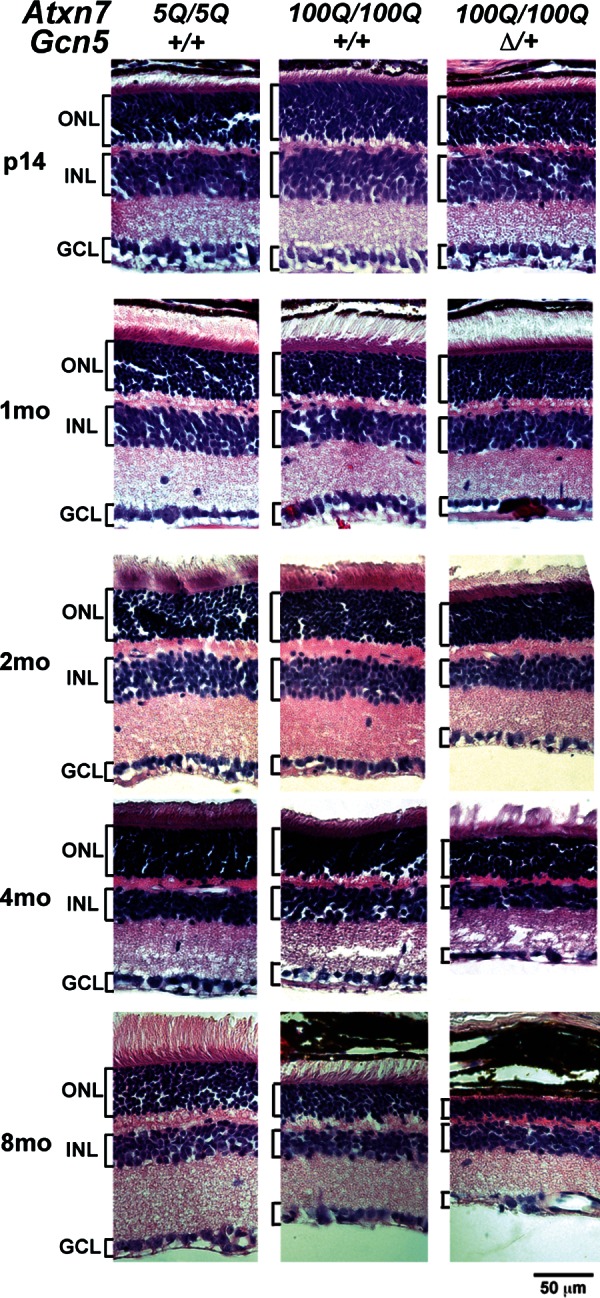
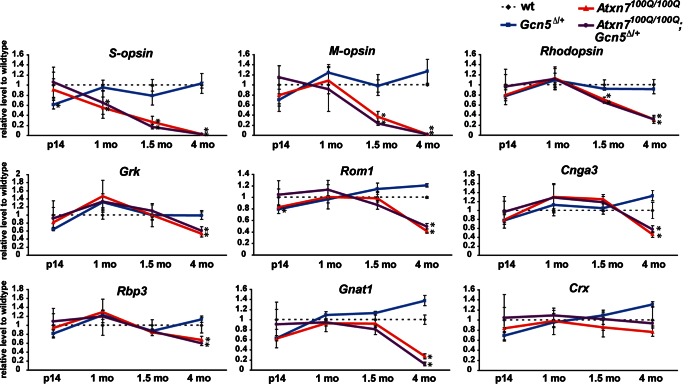
Spinocerebellar ataxia type 7 (SCA7) is a neurodegenerative disease caused by expansion of a CAG repeat encoding a polyglutamine tract in ATXN7, a component of the SAGA histone acetyltransferase (HAT) complex. Previous studies provided conflicting evidence regarding the effects of polyQ-ATXN7 on the activity of Gcn5, the HAT catalytic subunit of SAGA. Here, we report that reducing Gcn5 expression accelerates both cerebellar and retinal degeneration in a mouse model of SCA7. Deletion of Gcn5 in Purkinje cells in mice expressing wild-type (wt) Atxn7, however, causes only mild ataxia and does not lead to the early lethality observed in SCA7 mice. Reduced Gcn5 expression strongly enhances retinopathy in SCA7 mice, but does not affect the known transcriptional targets of Atxn7, as expression of these genes is not further altered by Gcn5 depletion. These findings demonstrate that loss of Gcn5 functions can contribute to the time of onset and severity of SCA7 phenotypes, and suggest that non-transcriptional functions of SAGA may play a role in neurodegeneration in this disease.
© The Author 2011. Published by Oxford University Press. All rights reserved.
Figures
References
-
- David G., Abbas N., Stevanin G., Durr A., Yvert G., Cancel G., Weber C., Imbert G., Saudou F., Antoniou E., et al. Cloning of the SCA7 gene reveals a highly unstable CAG repeat expansion. Nat. Genet. 1997;17:65–70. - PubMed
-
- David G., Durr A., Stevanin G., Cancel G., Abbas N., Benomar A., Belal S., Lebre A.-S., Abada-Bendib M., Grid D., et al. Molecular and clinical correlations in autosomal dominant cerebellar ataxia with progressive macular dystrophy (SCA7) Hum. Mol. Genet. 1998;7:165–170. - PubMed
-
- Del-Favero J., Krols L., Michalik A., Theuns J., Löfgren A., Goossens D., Wehnert A., Van den Bossche D., Van Zand K., Backhovens H., et al. Molecular genetic analysis of autosomal dominant cerebellar ataxia with retinal degeneration (ADCA Type II) caused by CAG triplet repeat expansion. Hum. Mol. Genet. 1998;7:177–186. - PubMed
-
- Johansson J., Forsgren L., Sandgren O., Brice A., Holmgren G., Holmberg M. Expanded CAG repeats in Swedish spinocerebellar ataxia type 7 (SCA7) patients: effect of CAG repeat length on the clinical manifestation. Hum. Mol. Genet. 1998;7:171–176. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
