

Normalization, testing, and false discovery rate estimation for RNA-sequencing data
- PMID: 22003245
- PMCID: PMC3372940
- DOI: 10.1093/biostatistics/kxr031
Normalization, testing, and false discovery rate estimation for RNA-sequencing data
Abstract
We discuss the identification of genes that are associated with an outcome in RNA sequencing and other sequence-based comparative genomic experiments. RNA-sequencing data take the form of counts, so models based on the Gaussian distribution are unsuitable. Moreover, normalization is challenging because different sequencing experiments may generate quite different total numbers of reads. To overcome these difficulties, we use a log-linear model with a new approach to normalization. We derive a novel procedure to estimate the false discovery rate (FDR). Our method can be applied to data with quantitative, two-class, or multiple-class outcomes, and the computation is fast even for large data sets. We study the accuracy of our approaches for significance calculation and FDR estimation, and we demonstrate that our method has potential advantages over existing methods that are based on a Poisson or negative binomial model. In summary, this work provides a pipeline for the significance analysis of sequencing data.
Figures
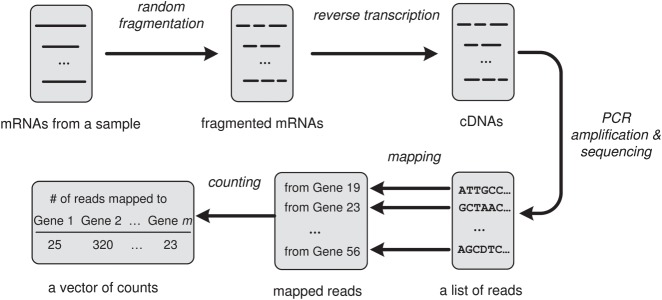


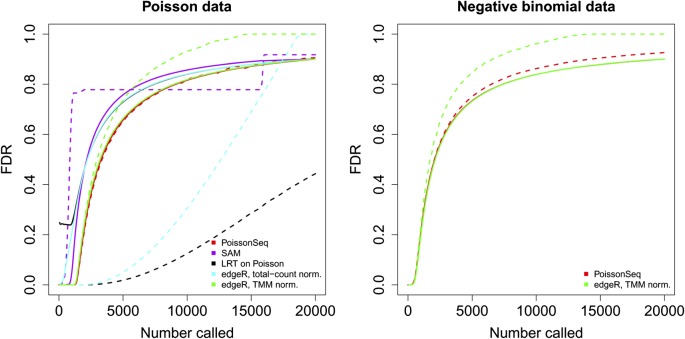
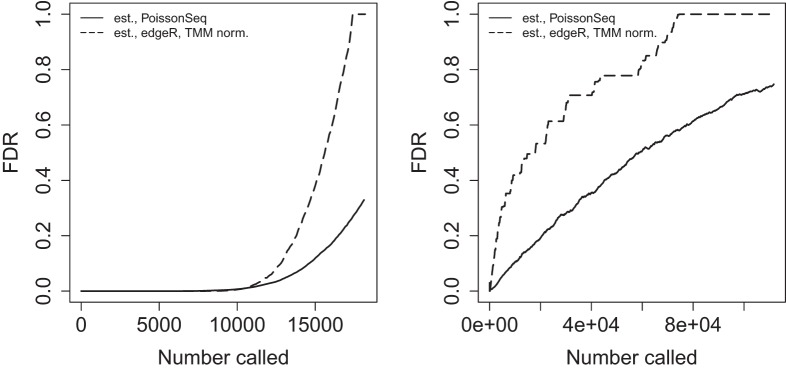
References
-
- Agresti A. Categorical Data Analysis. 2nd edition. New York: Wiley; 2002.
-
- Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society Series B. 1995;85:289–300.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
