

Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid
- PMID: 22008187
- PMCID: PMC3219791
- DOI: 10.1186/1471-2164-12-514
Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid
Abstract
Background: Higher plants exhibit remarkable phenotypic plasticity allowing them to adapt to an extensive range of environmental conditions. Sorghum is a cereal crop that exhibits exceptional tolerance to adverse conditions, in particular, water-limiting environments. This study utilized next generation sequencing (NGS) technology to examine the transcriptome of sorghum plants challenged with osmotic stress and exogenous abscisic acid (ABA) in order to elucidate genes and gene networks that contribute to sorghum's tolerance to water-limiting environments with a long-term aim of developing strategies to improve plant productivity under drought.
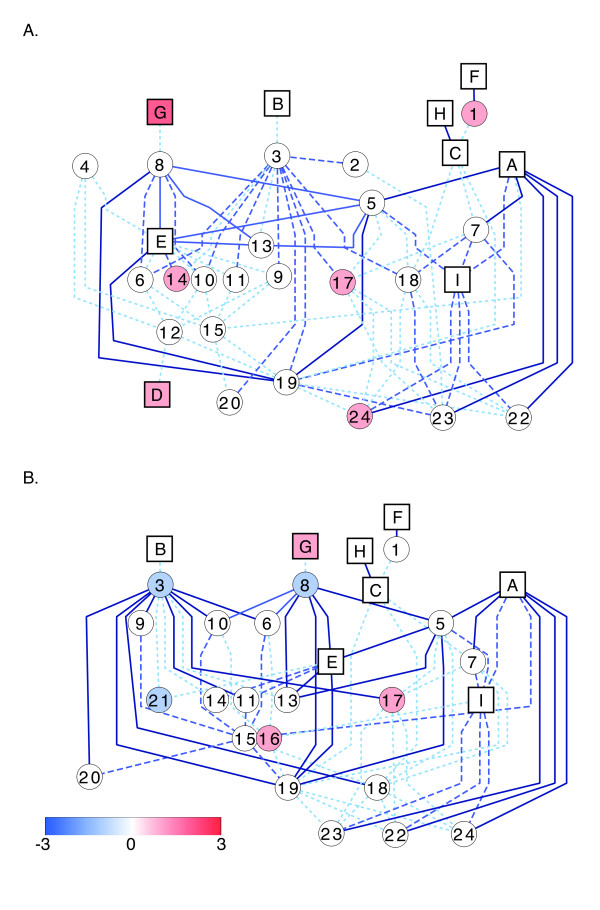
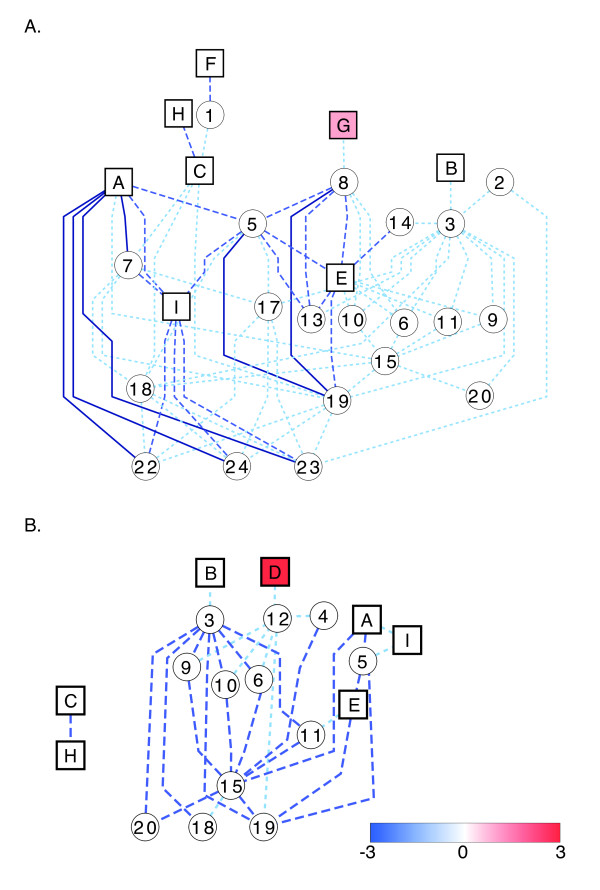
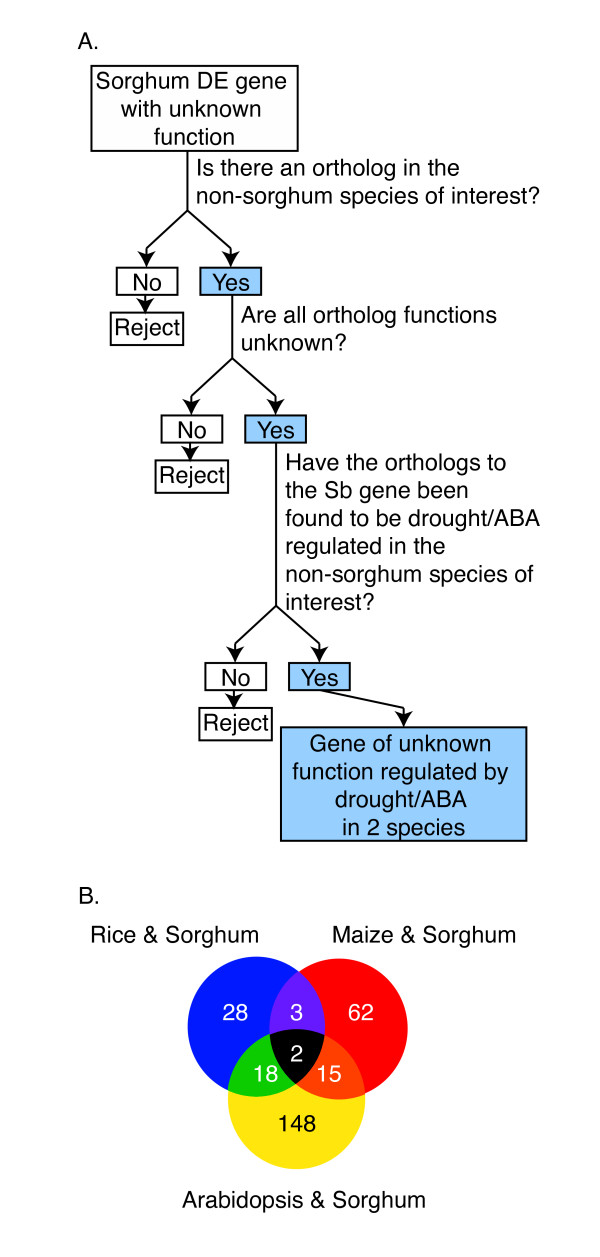
Results: RNA-Seq results revealed transcriptional activity of 28,335 unique genes from sorghum root and shoot tissues subjected to polyethylene glycol (PEG)-induced osmotic stress or exogenous ABA. Differential gene expression analyses in response to osmotic stress and ABA revealed a strong interplay among various metabolic pathways including abscisic acid and 13-lipoxygenase, salicylic acid, jasmonic acid, and plant defense pathways. Transcription factor analysis indicated that groups of genes may be co-regulated by similar regulatory sequences to which the expressed transcription factors bind. We successfully exploited the data presented here in conjunction with published transcriptome analyses for rice, maize, and Arabidopsis to discover more than 50 differentially expressed, drought-responsive gene orthologs for which no function had been previously ascribed.
Conclusions: The present study provides an initial assemblage of sorghum genes and gene networks regulated by osmotic stress and hormonal treatment. We are providing an RNA-Seq data set and an initial collection of transcription factors, which offer a preliminary look into the cascade of global gene expression patterns that arise in a drought tolerant crop subjected to abiotic stress. These resources will allow scientists to query gene expression and functional annotation in response to drought.
Figures
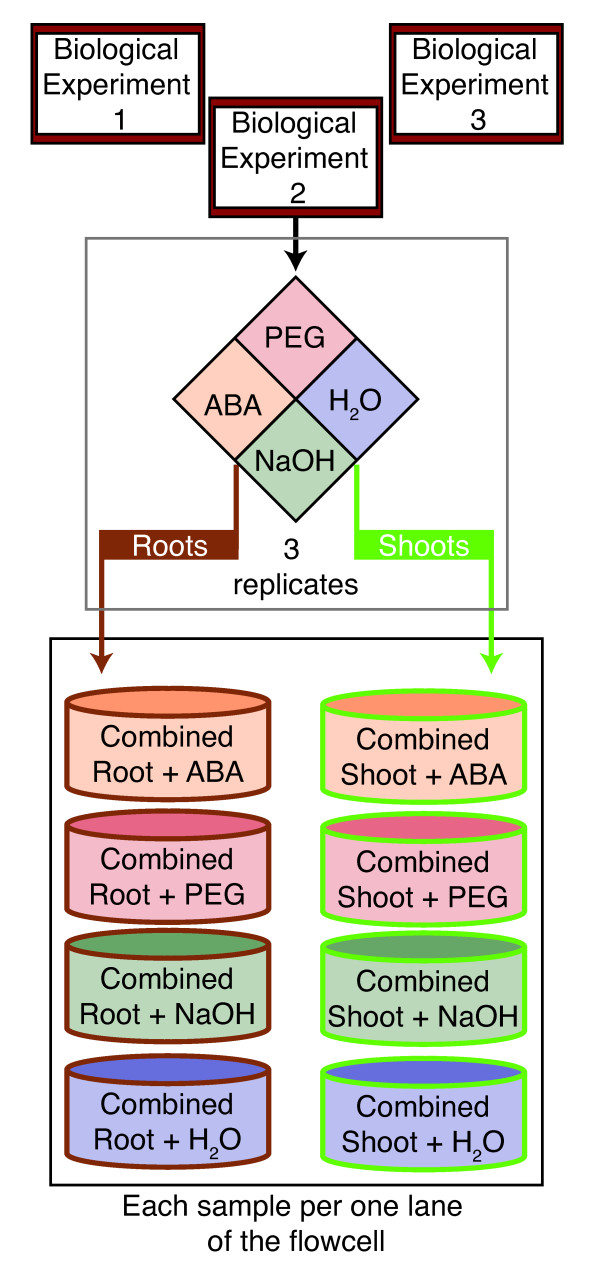
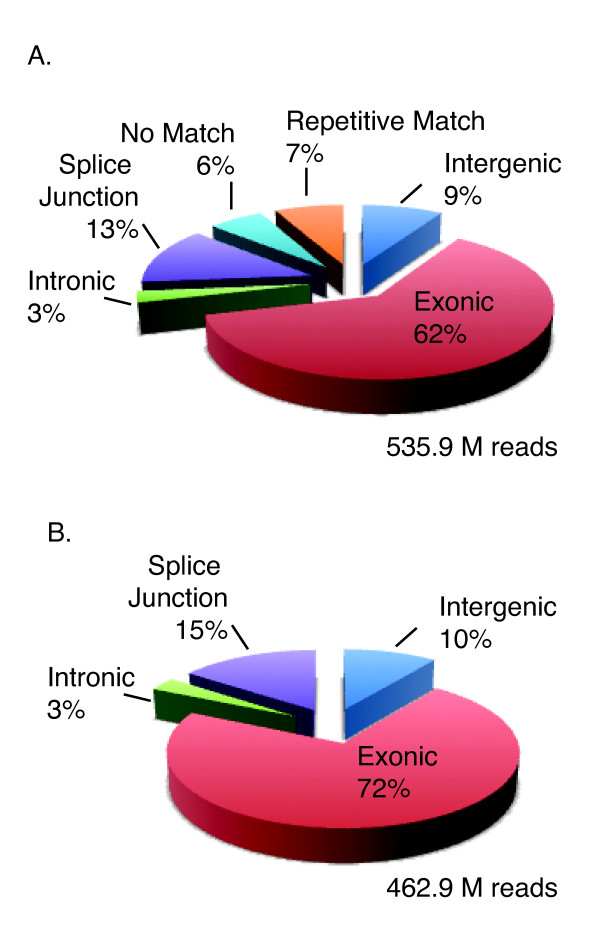
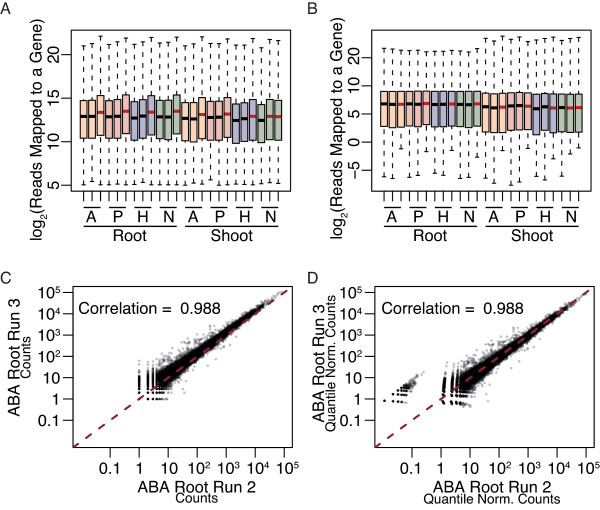
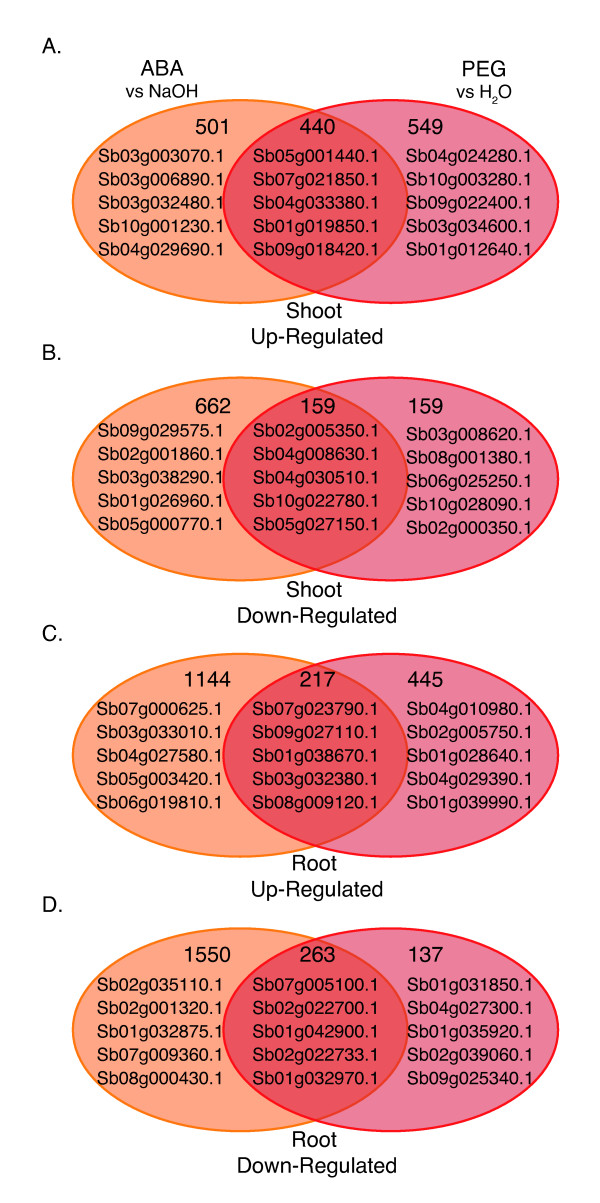
References
-
- Boyer JS. Plant productivity and environment. Science. 1982;218(4571):443–448. - PubMed
-
- Gleick PH. Global freshwater resources: soft-path solutions for the 21st century. Science. 2003;302(5650):1524–1528. - PubMed
-
- Johnson N, Revenga C, Echeverria J. Ecology. Managing water for people and nature. Science. 2001;292(5519):1071–1072. - PubMed
-
- Amber D. Genetic responses to drought. Scientist. 2000. pp. 18–19.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
