

Ocular manifestations in the Hutchinson-Gilford progeria syndrome
- PMID: 22011502
- PMCID: PMC3214428
- DOI: 10.4103/0301-4738.86327
Ocular manifestations in the Hutchinson-Gilford progeria syndrome
Abstract
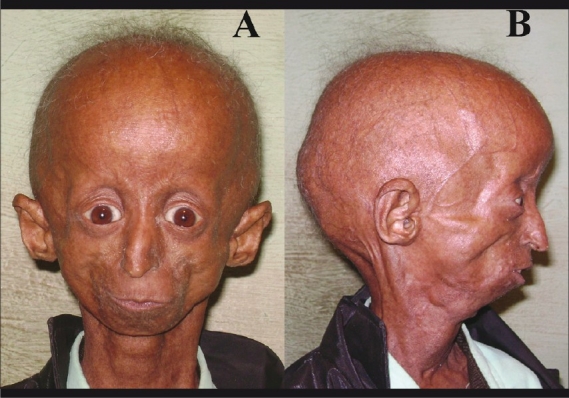
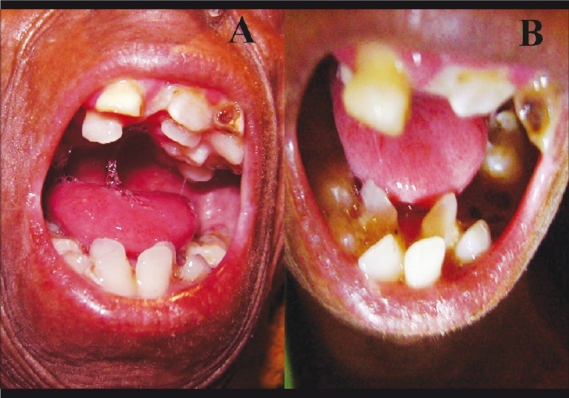
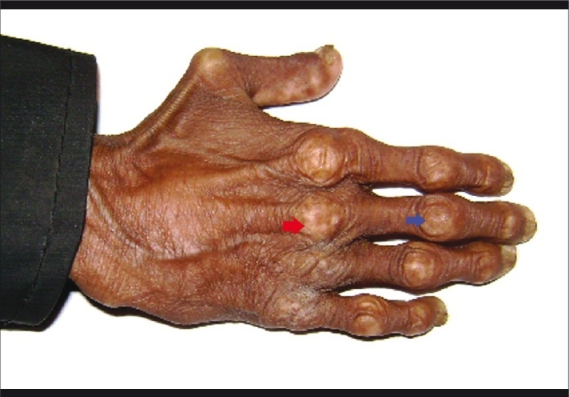
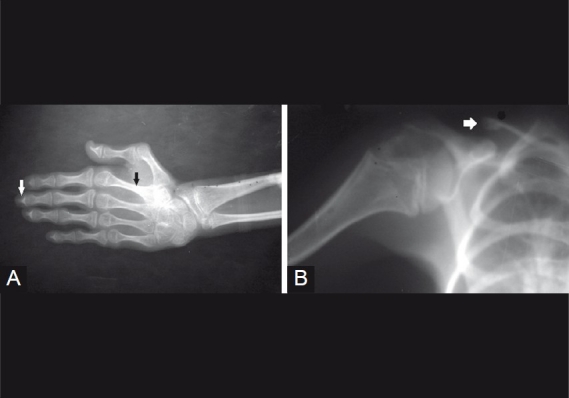
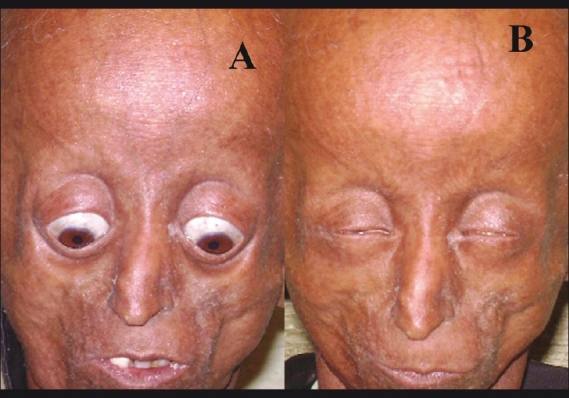
The Hutchinson-Gilford progeria (HGP) syndrome is an extremely rare genetic condition characterized by an appearance of accelerated aging in children. The word progeria is derived from the Greek word progeros meaning 'prematurely old'. It is caused by de novo dominant mutation in the LMNA gene (gene map locus 1q21.2) and characterized by growth retardation and accelerated degenerative changes of the skin, musculoskeletal and cardiovascular systems. The most common ocular manifestations are prominent eyes, loss of eyebrows and eyelashes, and lagophthalmos. In the present case some additional ocular features such as horizontal narrowing of palpebral fissure, superior sulcus deformity, upper lid retraction, upper lid lag in down gaze, poor pupillary dilatation, were noted. In this case report, a 15-year-old Indian boy with some additional ocular manifestations of the HGP syndrome is described.
Figures
Similar articles
-
Hutchinson-Gilford progeria syndrome.Indian J Dermatol Venereol Leprol. 2010 Sep-Oct;76(5):591. doi: 10.4103/0378-6323.69094. Indian J Dermatol Venereol Leprol. 2010. PMID: 20827016
-
Ocular manifestation in progeria: A case report.Nepal J Ophthalmol. 2020 Jan;12(23):113-138. doi: 10.3126/nepjoph.v12i1.25454. Nepal J Ophthalmol. 2020. PMID: 32799251
-
Hutchinson-Gilford progeria syndrome caused by an LMNA mutation: a case report.Pediatr Dermatol. 2015 Mar-Apr;32(2):271-5. doi: 10.1111/pde.12406. Epub 2014 Dec 29. Pediatr Dermatol. 2015. PMID: 25556323 Review.
-
Hutchinson-Gilford progeria syndrome: oral and craniofacial phenotypes.Oral Dis. 2009 Apr;15(3):187-95. doi: 10.1111/j.1601-0825.2009.01521.x. Epub 2009 Feb 19. Oral Dis. 2009. PMID: 19236595 Free PMC article.
-
Progeria infantum (Hutchinson-Gilford syndrome) associated with scleroderma-like lesions and acro-osteolysis: a case report and brief review of the literature.Pediatr Dermatol. 2000 Jul-Aug;17(4):282-5. doi: 10.1046/j.1525-1470.2000.01775.x. Pediatr Dermatol. 2000. PMID: 10990576 Review.
Cited by
-
Computational Exploration for Lead Compounds That Can Reverse the Nuclear Morphology in Progeria.Biomed Res Int. 2017;2017:5270940. doi: 10.1155/2017/5270940. Epub 2017 Oct 26. Biomed Res Int. 2017. PMID: 29226142 Free PMC article.
-
Progerin mRNA Is Associated with Smoking and Signs of Increased Microvascular Damage in Patients with Diabetic Macular Edema.Int J Mol Sci. 2025 Feb 27;26(5):2099. doi: 10.3390/ijms26052099. Int J Mol Sci. 2025. PMID: 40076719 Free PMC article.
-
Progeria Presenting with Pyogenic Granuloma in Conjunctiva: A Case Report.JNMA J Nepal Med Assoc. 2023 Jul;61(263):608-610. doi: 10.31729/jnma.8218. Epub 2023 Jul 30. JNMA J Nepal Med Assoc. 2023. PMID: 40802724 Free PMC article.
References
-
- Hennekam RC. Hutchinson-Gilford progeria syndrome: Review of the phenotype. Am J Med Genet A. 2006;140:2603–24. - PubMed
-
- Brown TW. Progeria. In: Kliegman RM, Bohrman RE, Jenson HB, Stanton BF, editors. Nelson Textbook of Pediatrics. 18th ed. Philadelphia: Saunders Elsevier; 2007. pp. 636–7.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
