

Post-burn hepatic insulin resistance is associated with endoplasmic reticulum (ER) stress
- PMID: 22011639
- PMCID: PMC3876730
- DOI: 10.1097/SHK.0b013e3181b2f439
Post-burn hepatic insulin resistance is associated with endoplasmic reticulum (ER) stress
Abstract
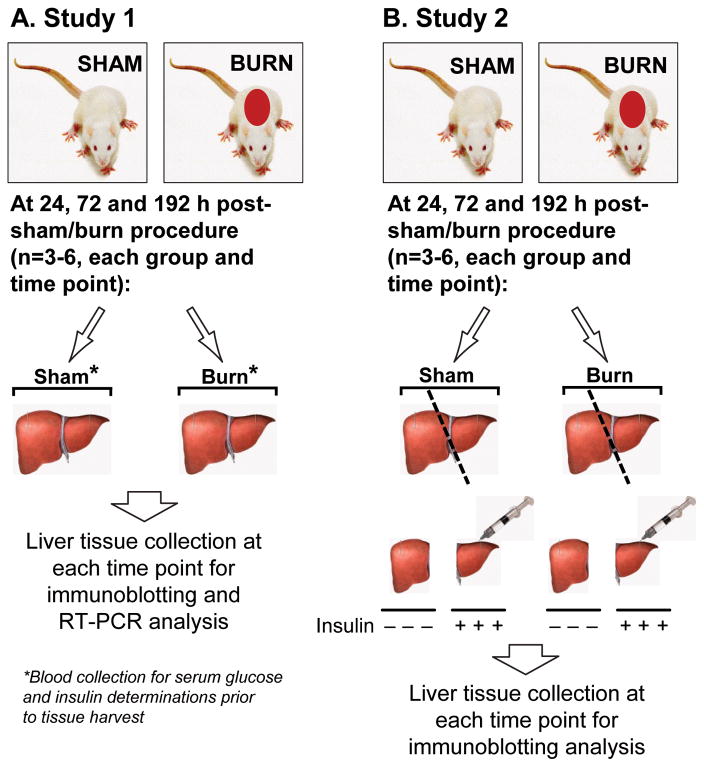
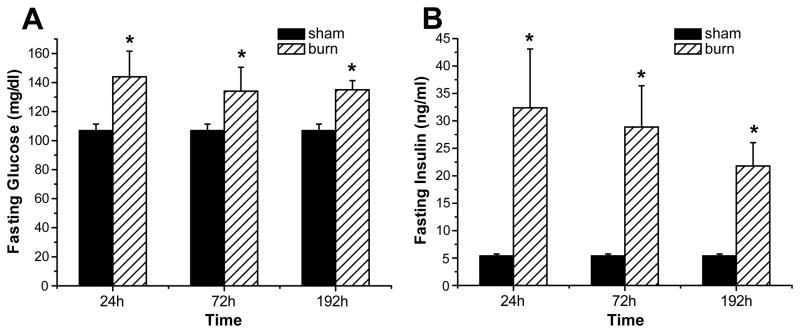
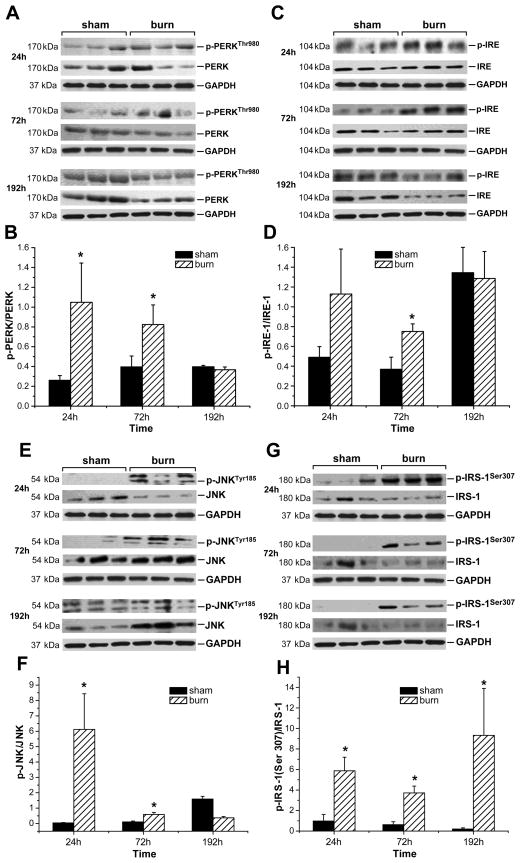
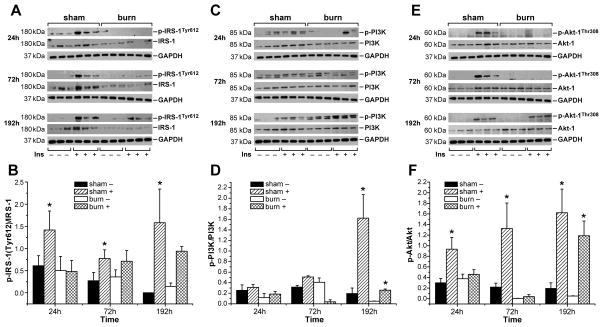
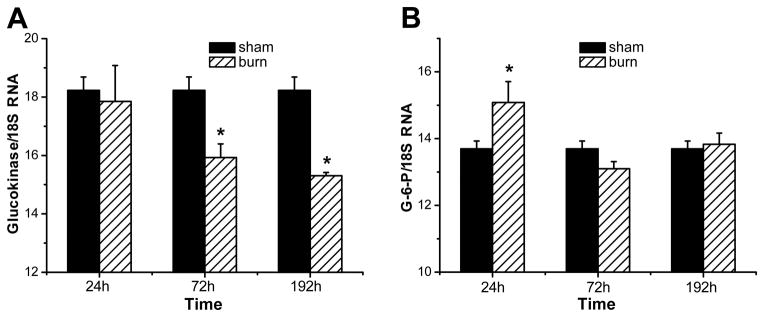
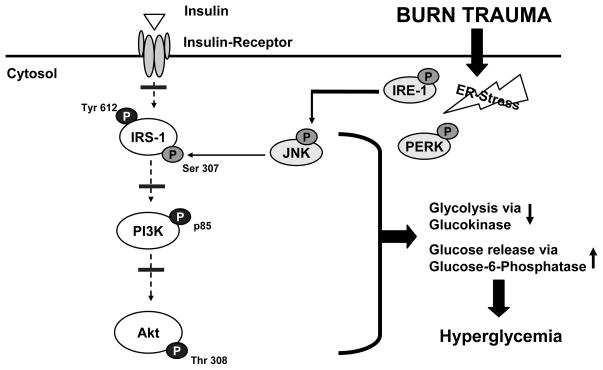
Insulin resistance with its associated hyperglycemias represents one significant contributor to mortality in burned patients. A variety of cellular stress-signaling pathways are activated as a consequence of burn. A key player in the cellular stress response is the endoplasmic reticulum (ER). Here, we investigated a possible role for ER-stress pathways in the progression of insulin function dysregulation postburn. Rats received a 60% total body surface area thermal injury, and a laparotomy was performed at 24, 72, and 192 h postburn. Liver was harvested before and 1 min after insulin injection (1 IU/kg) into the portal vein, and expression patterns of various proteins known to be involved in insulin and ER-stress signaling were determined by Western blotting. mRNA expression of glucose-6-phosphatase and glucokinase were determined by reverse-transcriptase-polymerase chain reaction and fasting serum glucose and insulin levels by standard enzymatic and enzyme-linked immunosorbent assay techniques, respectively. Insulin resistance indicated by increased glucose and insulin levels occurred starting 24 h postburn. Burn injury resulted in activation of ER stress pathways, reflected by significantly increased accumulation of phospho-PKR-like ER-kinase and phosphorylated inositol requiring enzyme 1, leading to an elevation of phospho-c-Jun N-terminal kinase and serine phosphorylation of insulin receptor substrate (IRS) 1 postburn. Insulin administration caused a significant increase in tyrosine phosphorylation of IRS-1, leading to activation of the phosphatidylinositol 3 kinase/Akt pathway in normal liver. Postburn tyrosine phosphorylation of IRS-1 was significantly impaired, associated with an inactivation of signaling molecules acting downstream of IRS-1, leading to significantly elevated transcription of glucose-6-phosphatase and significantly decreased mRNA expression of glucokinase. Activation of ER-stress signaling cascades may explain metabolic abnormalities involving insulin action after burn.
Figures
Similar articles
-
Propranolol improves impaired hepatic phosphatidylinositol 3-kinase/akt signaling after burn injury.Mol Med. 2012 May 9;18(1):707-11. doi: 10.2119/molmed.2011.00277. Mol Med. 2012. PMID: 22396018 Free PMC article.
-
Dissociation of inositol-requiring enzyme (IRE1α)-mediated c-Jun N-terminal kinase activation from hepatic insulin resistance in conditional X-box-binding protein-1 (XBP1) knock-out mice.J Biol Chem. 2012 Jan 20;287(4):2558-67. doi: 10.1074/jbc.M111.316760. Epub 2011 Nov 28. J Biol Chem. 2012. PMID: 22128176 Free PMC article.
-
Molecular mechanism(s) of burn-induced insulin resistance in murine skeletal muscle: role of IRS phosphorylation.Life Sci. 2005 Oct 28;77(24):3068-77. doi: 10.1016/j.lfs.2005.02.034. Epub 2005 Jun 27. Life Sci. 2005. PMID: 15982669
-
Endoplasmic reticulum stress and insulin resistance post-trauma: similarities to type 2 diabetes.J Cell Mol Med. 2012 Mar;16(3):437-44. doi: 10.1111/j.1582-4934.2011.01405.x. J Cell Mol Med. 2012. PMID: 21812914 Free PMC article. Review.
-
Drugs Interfering with Insulin Resistance and Their Influence on the Associated Hypermetabolic State in Severe Burns: A Narrative Review.Int J Mol Sci. 2021 Sep 10;22(18):9782. doi: 10.3390/ijms22189782. Int J Mol Sci. 2021. PMID: 34575946 Free PMC article. Review.
Cited by
-
Accumulation of myeloid lineage cells is mapping out liver fibrosis post injury: a targetable lesion using Ketanserin.Exp Mol Med. 2018 Jul 19;50(7):1-13. doi: 10.1038/s12276-018-0118-x. Exp Mol Med. 2018. PMID: 30026607 Free PMC article.
-
Role of Endoplasmic Reticulum Stress-Autophagy Axis in Severe Burn-Induced Intestinal Tight Junction Barrier Dysfunction in Mice.Front Physiol. 2019 May 22;10:606. doi: 10.3389/fphys.2019.00606. eCollection 2019. Front Physiol. 2019. PMID: 31191335 Free PMC article.
-
The biochemical alterations underlying post-burn hypermetabolism.Biochim Biophys Acta Mol Basis Dis. 2017 Oct;1863(10 Pt B):2633-2644. doi: 10.1016/j.bbadis.2017.02.019. Epub 2017 Feb 20. Biochim Biophys Acta Mol Basis Dis. 2017. PMID: 28219767 Free PMC article. Review.
-
Statistical and clinical analysis of alterations in glucose values after burns.Ann Burns Fire Disasters. 2016 Sep 30;29(3):163-171. Ann Burns Fire Disasters. 2016. PMID: 28149243 Free PMC article.
-
Pathophysiological Response to Burn Injury in Adults.Ann Surg. 2018 Mar;267(3):576-584. doi: 10.1097/SLA.0000000000002097. Ann Surg. 2018. PMID: 29408836 Free PMC article.
References
-
- Thombs BD, Singh VA, Milner SM. Children under 4 years are at greater risk of mortality following acute burn injury: evidence from a national sample of 12,902 pediatric admissions. Shock. 2006;26:348–352. - PubMed
-
- Lorente JA, Vallejo A, Galeiras R, Tomicic V, Zamora J, Cerda E, de la Cal MA, Esteban A. Organ dysfunction as estimated by the sequential organ failure assessment score is related to outcome in critically ill burn patients. Shock. 2009;31:125–131. - PubMed
-
- Herndon DN, Tompkins RG. Support of the metabolic response to burn injury. Lancet. 2004;363:1895–1902. - PubMed
-
- McCowen KC, Malhotra A, Bistrian BR. Stress-induced hyperglycemia. Crit Care Clin. 2001;17:107–124. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
