

Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis
- PMID: 22013014
- PMCID: PMC3219956
- DOI: 10.2337/db11-0061
Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis
Abstract
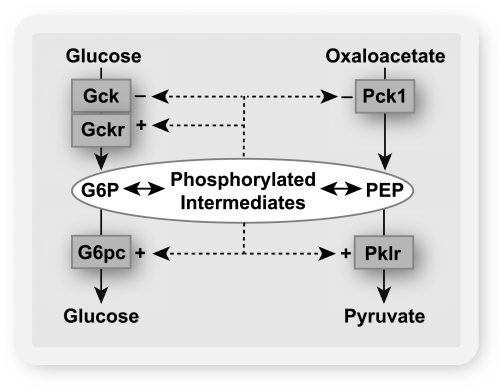
Objective: The induction of hepatic glucose 6-phosphatase (G6pc) by glucose presents a paradox of glucose-induced glucose intolerance. We tested whether glucose regulation of liver gene expression is geared toward intracellular homeostasis.
Research design and methods: The effect of glucose-induced accumulation of phosphorylated intermediates on expression of glucokinase (Gck) and its regulator Gckr was determined in hepatocytes. Cell ATP and uric acid production were measured as indices of cell phosphate homeostasis.
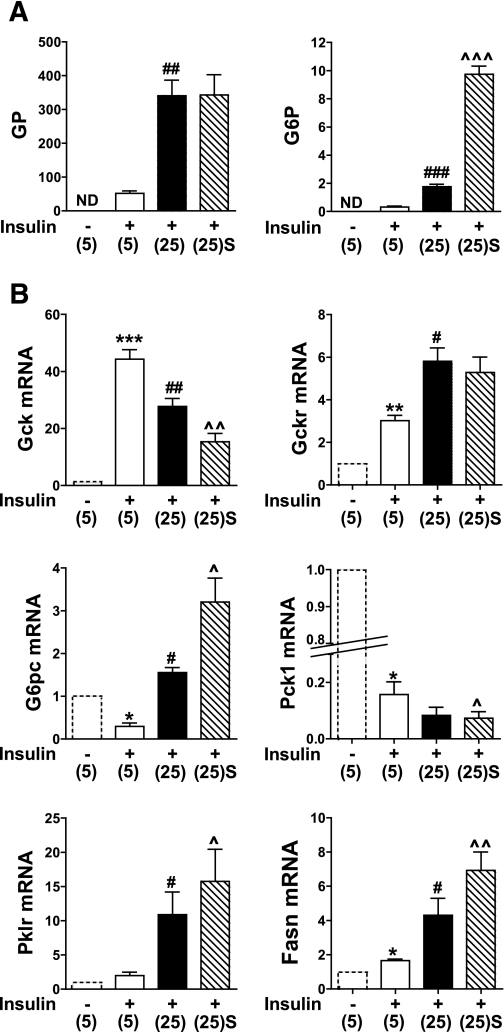
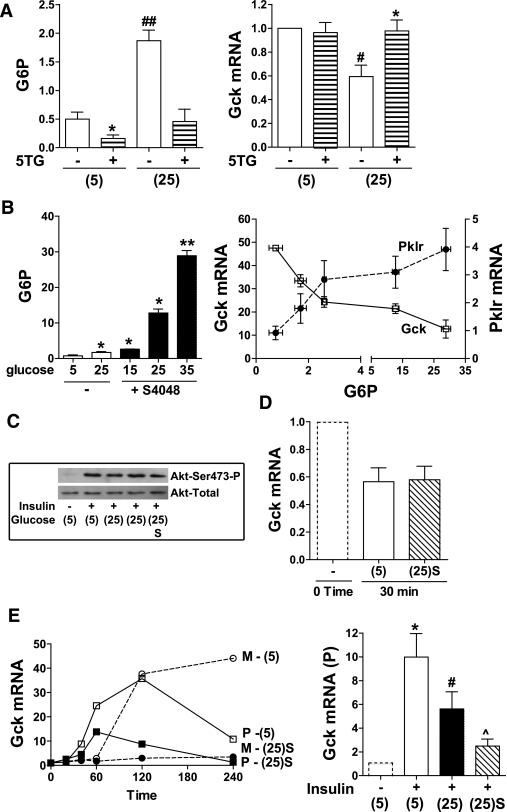
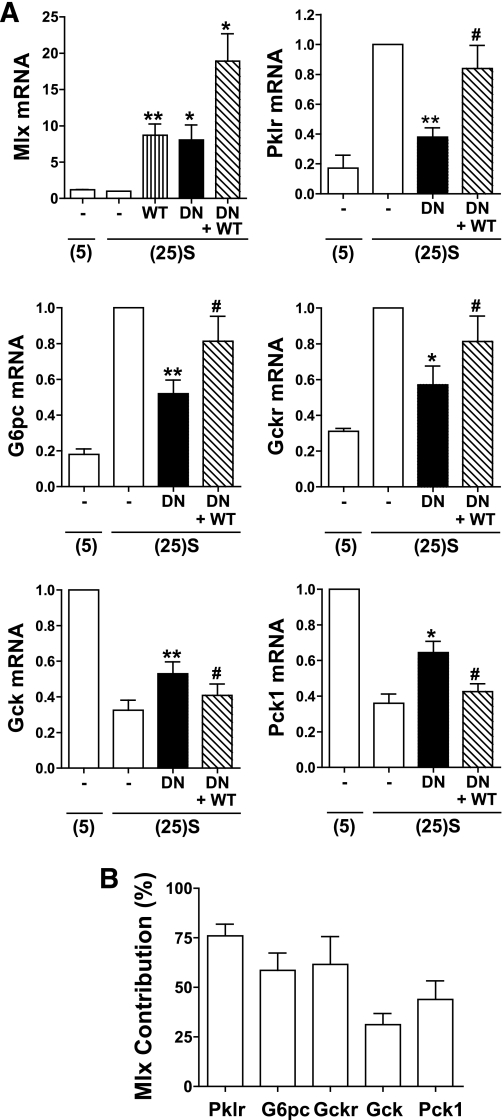
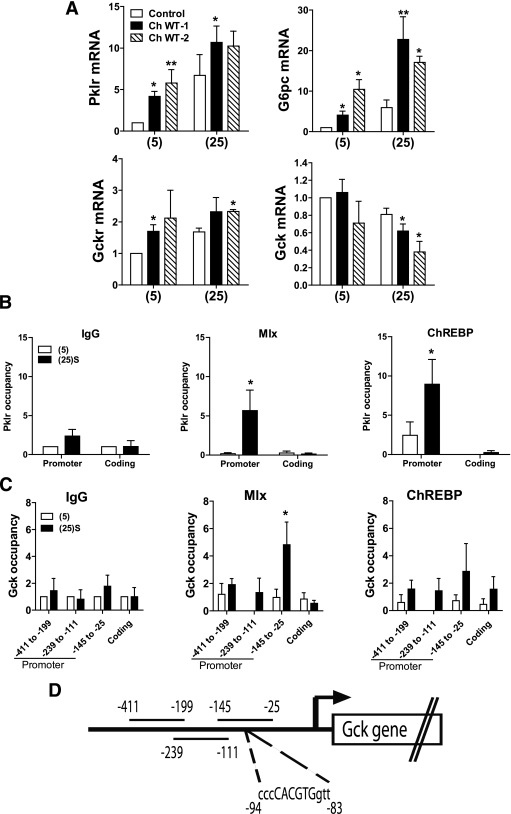
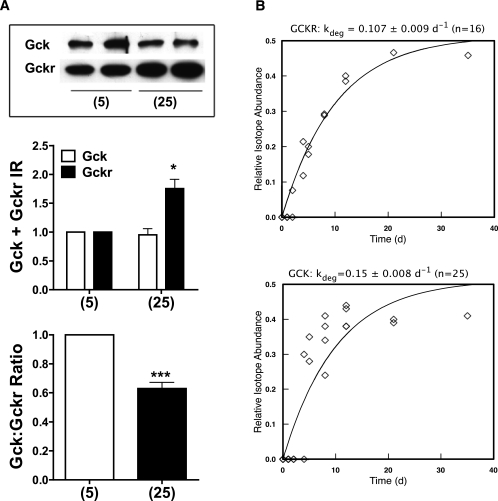
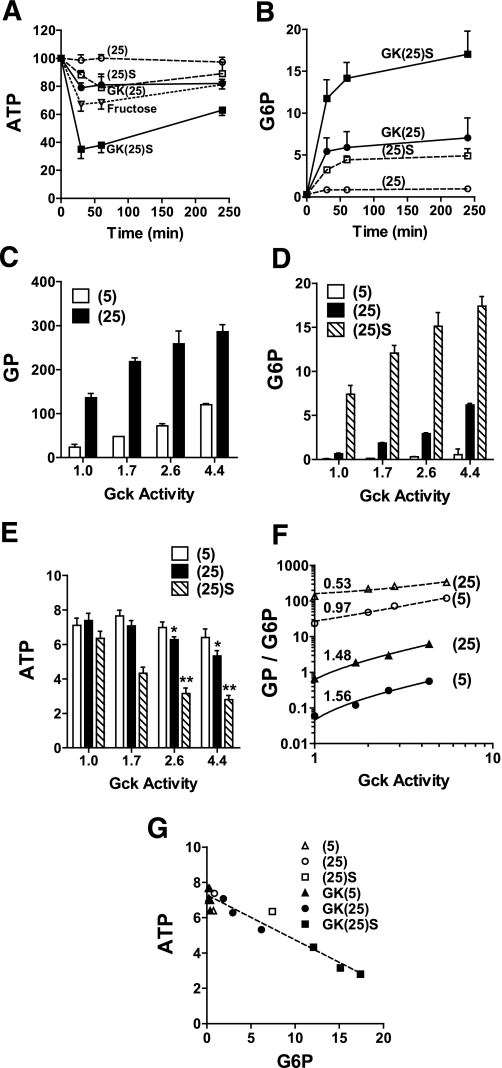
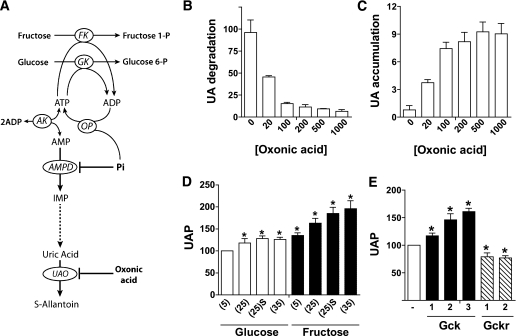
Results: Accumulation of phosphorylated intermediates in hepatocytes incubated at elevated glucose induced rapid and inverse changes in Gck (repression) and Gckr (induction) mRNA concomitantly with induction of G6pc, but had slower effects on the Gckr-to-Gck protein ratio. Dynamic metabolic labeling in mice and liver proteome analysis confirmed that Gckr and Gck are low-turnover proteins. Involvement of Max-like protein X in glucose-mediated Gck-repression was confirmed by chromatin immunoprecipitation analysis. Elevation of the Gck-to-Gckr ratio in hepatocytes was associated with glucose-dependent ATP depletion and elevated urate production confirming compromised phosphate homeostasis.
Conclusions: The lowering by glucose of the Gck-to-Gckr ratio provides a potential explanation for the impaired hepatic glucose uptake in diabetes. Elevated uric acid production at an elevated Gck-to-Gckr ratio supports a role for glucose regulation of gene expression in hepatic phosphate homeostasis.
Figures
Similar articles
-
Opposite effects of a glucokinase activator and metformin on glucose-regulated gene expression in hepatocytes.Diabetes Obes Metab. 2017 Aug;19(8):1078-1087. doi: 10.1111/dom.12910. Epub 2017 Apr 10. Diabetes Obes Metab. 2017. PMID: 28206714
-
High-carbohydrate diets induce hepatic insulin resistance to protect the liver from substrate overload.Biochem Pharmacol. 2013 Feb 1;85(3):306-12. doi: 10.1016/j.bcp.2012.09.019. Epub 2012 Sep 26. Biochem Pharmacol. 2013. PMID: 23022226
-
Cellular characterisation of the GCKR P446L variant associated with type 2 diabetes risk.Diabetologia. 2012 Jan;55(1):114-22. doi: 10.1007/s00125-011-2348-5. Epub 2011 Oct 25. Diabetologia. 2012. PMID: 22038520 Free PMC article.
-
The Protective Role of the Carbohydrate Response Element Binding Protein in the Liver: The Metabolite Perspective.Front Endocrinol (Lausanne). 2020 Nov 17;11:594041. doi: 10.3389/fendo.2020.594041. eCollection 2020. Front Endocrinol (Lausanne). 2020. PMID: 33281747 Free PMC article. Review.
-
Evolution of glucose utilization: glucokinase and glucokinase regulator protein.Mol Phylogenet Evol. 2014 Jan;70:195-203. doi: 10.1016/j.ympev.2013.09.016. Epub 2013 Sep 25. Mol Phylogenet Evol. 2014. PMID: 24075984 Free PMC article. Review.
Cited by
-
Adiposity amplifies the genetic risk of fatty liver disease conferred by multiple loci.Nat Genet. 2017 Jun;49(6):842-847. doi: 10.1038/ng.3855. Epub 2017 Apr 24. Nat Genet. 2017. PMID: 28436986 Free PMC article.
-
PAS Kinase deficiency alters the glucokinase function and hepatic metabolism.Sci Rep. 2018 Jul 23;8(1):11091. doi: 10.1038/s41598-018-29234-8. Sci Rep. 2018. PMID: 30038292 Free PMC article.
-
ChREBP is activated by reductive stress and mediates GCKR-associated metabolic traits.Cell Metab. 2024 Jan 2;36(1):144-158.e7. doi: 10.1016/j.cmet.2023.11.010. Epub 2023 Dec 14. Cell Metab. 2024. PMID: 38101397 Free PMC article.
-
Exendin-4 stimulates autophagy in pancreatic β-cells via the RAPGEF/EPAC-Ca2+-PPP3/calcineurin-TFEB axis.Autophagy. 2022 Apr;18(4):799-815. doi: 10.1080/15548627.2021.1956123. Epub 2021 Aug 2. Autophagy. 2022. PMID: 34338148 Free PMC article.
-
Nuclear receptor corepressors non-canonically drive glucocorticoid receptor-dependent activation of hepatic gluconeogenesis.Nat Metab. 2024 May;6(5):825-836. doi: 10.1038/s42255-024-01029-4. Epub 2024 Apr 15. Nat Metab. 2024. PMID: 38622413 Free PMC article.
References
-
- Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J 2008;414:1–18 - PubMed
-
- Agius L, Peak M, Newgard CB, Gomez-Foix AM, Guinovart JJ. Evidence for a role of glucose-induced translocation of glucokinase in the control of hepatic glycogen synthesis. J Biol Chem 1996;271:30479–30486 - PubMed
-
- Van Schaftingen E. Glycolysis revisited. Diabetologia 1993;36:581–588 - PubMed
-
- de la Iglesia N, Mukhtar M, Seoane J, Guinovart JJ, Agius L. The role of the regulatory protein of glucokinase in the glucose sensory mechanism of the hepatocyte. J Biol Chem 2000;275:10597–10603 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
