

Adeno-associated virus type 2 modulates the host DNA damage response induced by herpes simplex virus 1 during coinfection
- PMID: 22013059
- PMCID: PMC3255894
- DOI: 10.1128/JVI.05694-11
Adeno-associated virus type 2 modulates the host DNA damage response induced by herpes simplex virus 1 during coinfection
Abstract
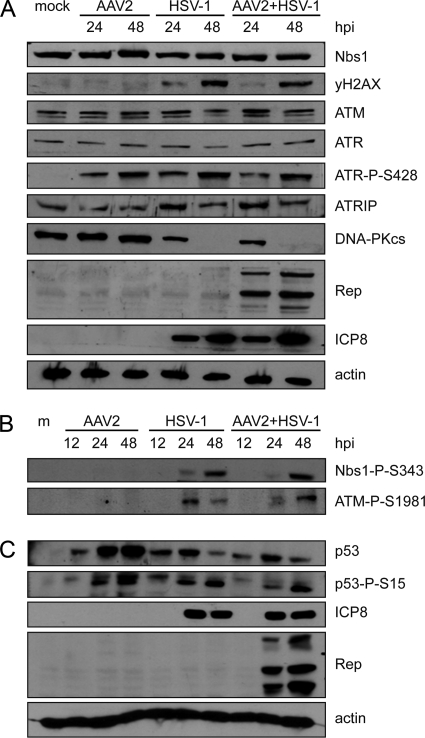
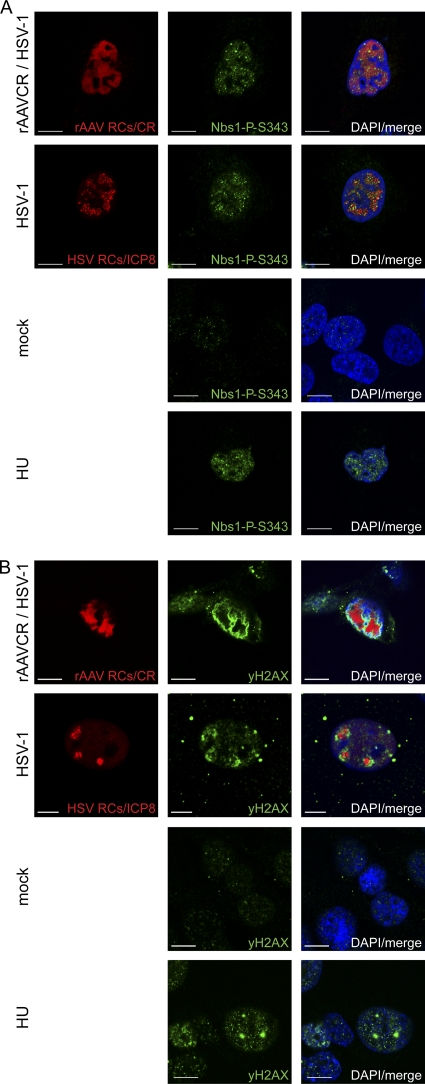
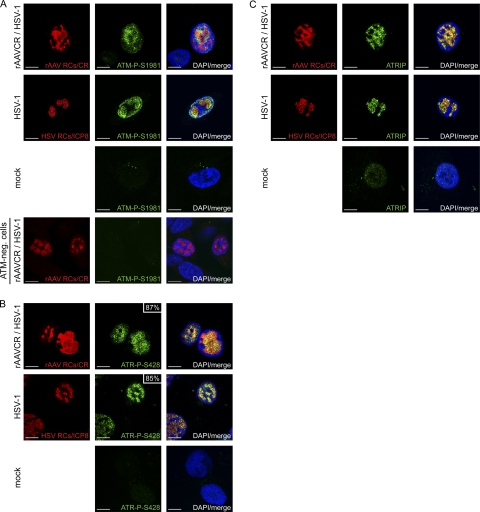
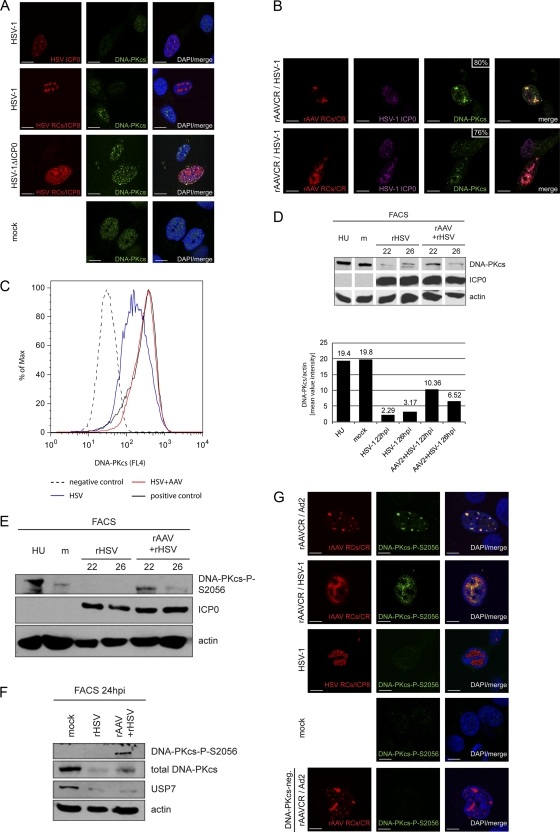
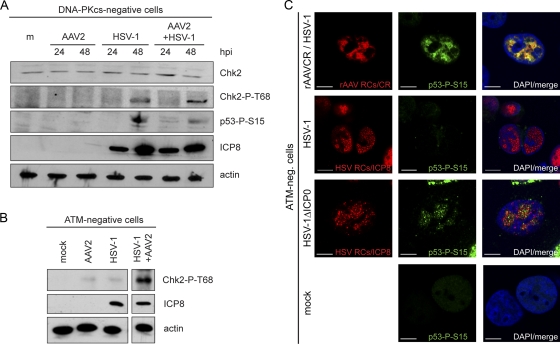
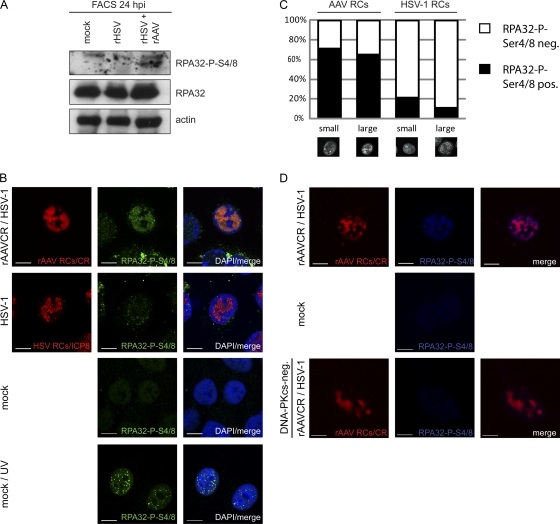
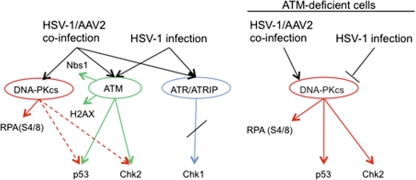
Adeno-associated virus type 2 (AAV2) is a human parvovirus that relies on a helper virus for efficient replication. Herpes simplex virus 1 (HSV-1) supplies helper functions and changes the environment of the cell to promote AAV2 replication. In this study, we examined the accumulation of cellular replication and repair proteins at viral replication compartments (RCs) and the influence of replicating AAV2 on HSV-1-induced DNA damage responses (DDR). We observed that the ATM kinase was activated in cells coinfected with AAV2 and HSV-1. We also found that phosphorylated ATR kinase and its cofactor ATR-interacting protein were recruited into AAV2 RCs, but ATR signaling was not activated. DNA-PKcs, another main kinase in the DDR, was degraded during HSV-1 infection in an ICP0-dependent manner, and this degradation was markedly delayed during AAV2 coinfection. Furthermore, we detected phosphorylation of DNA-PKcs during AAV2 but not HSV-1 replication. The AAV2-mediated delay in DNA-PKcs degradation affected signaling through downstream substrates. Overall, our results demonstrate that coinfection with HSV-1 and AAV2 provokes a cellular DDR which is distinct from that induced by HSV-1 alone.
Figures
References
-
- Ahn JY, Schwarz JK, Piwnica-Worms H, Canman CE. 2000. Threonine 68 phosphorylation by ataxia telangiectasia mutated is required for efficient activation of Chk2 in response to ionizing radiation. Cancer Res. 60:5934–5936 - PubMed
-
- Anantha RW, Vassin VM, Borowiec JA. 2007. Sequential and synergistic modification of human RPA stimulates chromosomal DNA repair. J. Biol. Chem. 282:35910–35923 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
