

Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif
- PMID: 22013910
- PMCID: PMC3209457
- DOI: 10.1186/1472-6807-11-43
Computational analysis of a novel mutation in ETFDH gene highlights its long-range effects on the FAD-binding motif
Abstract
Background: Multiple acyl-coenzyme A dehydrogenase deficiency (MADD) is an autosomal recessive disease caused by the defects in the mitochondrial electron transfer system and the metabolism of fatty acids. Recently, mutations in electron transfer flavoprotein dehydrogenase (ETFDH) gene, encoding electron transfer flavoprotein:ubiquinone oxidoreductase (ETF:QO) have been reported to be the major causes of riboflavin-responsive MADD. To date, no studies have been performed to explore the functional impact of these mutations or their mechanism of disrupting enzyme activity.
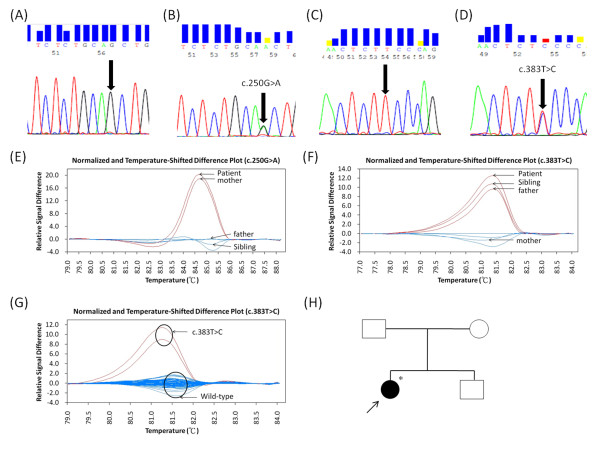
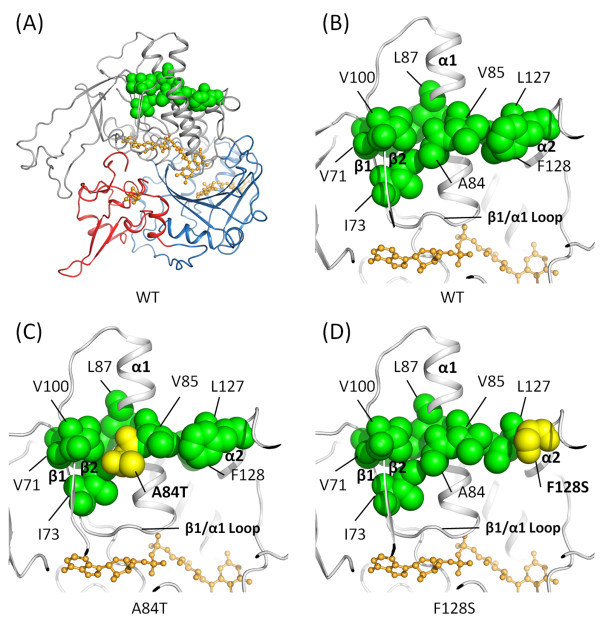
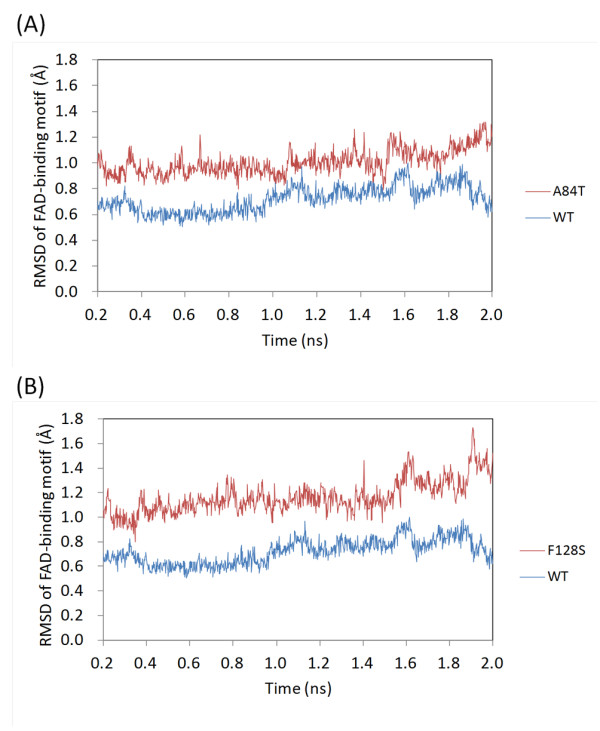
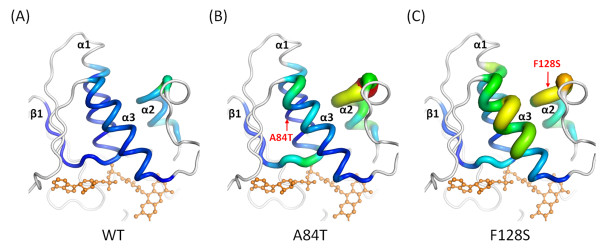
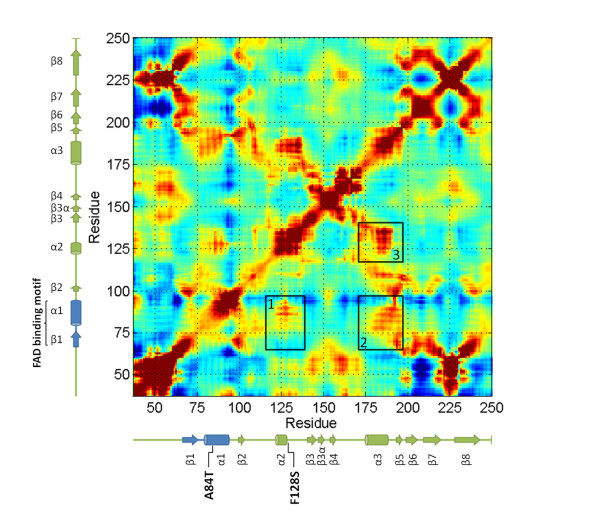
Results: High resolution melting (HRM) analysis and sequencing of the entire ETFDH gene revealed a novel mutation (p.Phe128Ser) and the hotspot mutation (p.Ala84Thr) from a patient with MADD. According to the predicted 3D structure of ETF:QO, the two mutations are located within the flavin adenine dinucleotide (FAD) binding domain; however, the two residues do not have direct interactions with the FAD ligand. Using molecular dynamics (MD) simulations and normal mode analysis (NMA), we found that the p.Ala84Thr and p.Phe128Ser mutations are most likely to alter the protein structure near the FAD binding site as well as disrupt the stability of the FAD binding required for the activation of ETF:QO. Intriguingly, NMA revealed that several reported disease-causing mutations in the ETF:QO protein show highly correlated motions with the FAD-binding site.
Conclusions: Based on the present findings, we conclude that the changes made to the amino acids in ETF:QO are likely to influence the FAD-binding stability.
Figures
Similar articles
-
ETFDH Mutations and Flavin Adenine Dinucleotide Homeostasis Disturbance Are Essential for Developing Riboflavin-Responsive Multiple Acyl-Coenzyme A Dehydrogenation Deficiency.Ann Neurol. 2018 Nov;84(5):659-673. doi: 10.1002/ana.25338. Epub 2018 Oct 19. Ann Neurol. 2018. PMID: 30232818
-
Molecular mechanisms of riboflavin responsiveness in patients with ETF-QO variations and multiple acyl-CoA dehydrogenation deficiency.Hum Mol Genet. 2012 Aug 1;21(15):3435-48. doi: 10.1093/hmg/dds175. Epub 2012 May 18. Hum Mol Genet. 2012. PMID: 22611163
-
The iron-sulfur cluster of electron transfer flavoprotein-ubiquinone oxidoreductase is the electron acceptor for electron transfer flavoprotein.Biochemistry. 2008 Aug 26;47(34):8894-901. doi: 10.1021/bi800507p. Epub 2008 Aug 2. Biochemistry. 2008. PMID: 18672901 Free PMC article.
-
ETF dehydrogenase advances in molecular genetics and impact on treatment.Crit Rev Biochem Mol Biol. 2021 Aug;56(4):360-372. doi: 10.1080/10409238.2021.1908952. Epub 2021 Apr 7. Crit Rev Biochem Mol Biol. 2021. PMID: 33823724 Review.
-
The electron transfer flavoprotein: ubiquinone oxidoreductases.Biochim Biophys Acta. 2010 Dec;1797(12):1910-6. doi: 10.1016/j.bbabio.2010.10.007. Epub 2010 Oct 16. Biochim Biophys Acta. 2010. PMID: 20937244 Review.
Cited by
-
Fatty Acid oxidation disorders in a chinese population in taiwan.JIMD Rep. 2013;11:165-72. doi: 10.1007/8904_2013_236. Epub 2013 May 23. JIMD Rep. 2013. PMID: 23700290 Free PMC article.
-
Multiple Acyl-CoA Dehydrogenation Deficiency (Glutaric Aciduria Type II) with a Novel Mutation of Electron Transfer Flavoprotein-Dehydrogenase in a Cat.JIMD Rep. 2014;13:43-51. doi: 10.1007/8904_2013_268. Epub 2013 Oct 20. JIMD Rep. 2014. PMID: 24142280 Free PMC article.
-
Multiple Acyl-CoA Dehydrogenase Deficiency with Variable Presentation Due to a Homozygous Mutation in a Bedouin Tribe.Genes (Basel). 2021 Jul 28;12(8):1140. doi: 10.3390/genes12081140. Genes (Basel). 2021. PMID: 34440319 Free PMC article.
-
Compound heterozygous mutations in electron transfer flavoprotein dehydrogenase identified in a young Chinese woman with late-onset glutaric aciduria type II.Lipids Health Dis. 2017 Sep 26;16(1):185. doi: 10.1186/s12944-017-0576-5. Lipids Health Dis. 2017. PMID: 28950901 Free PMC article.
-
Oxidative Phosphorylation System in Gastric Carcinomas and Gastritis.Oxid Med Cell Longev. 2017;2017:1320241. doi: 10.1155/2017/1320241. Epub 2017 Jun 28. Oxid Med Cell Longev. 2017. PMID: 28744336 Free PMC article. Clinical Trial.
References
-
- Frerman FE, Goodman SI. In: The metabolic and molecular bases of inherited disease. 8. Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editor. New York: McGraw-Hill; 2001. Defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric aciduria type II; pp. 2357–2365.
-
- Gregersen N, Kolvraa S, Rasmussen K, Christensen E, Brandt NJ, Ebbesen F, Hansen FH. Biochemical studies in a patient with defects in the metabolism of acyl-CoA and sarcosine: another possible case of glutaric aciduria type II. Journal of Inherited Metabolic Disease. 1981;3(3):67–72. - PubMed
-
- Olsen RKJ, Olpin SE, Andresen BS, Miedzybrodzka ZH, Pourfarzam M, Merinero B, Frerman FE, Beresford MW, Dean JCS, Cornelius N. et al.ETFDH mutations as a major cause of riboflavin-responsive multiple acyl-CoA dehydrogenation deficiency. Brain. 2007;130:2045–2054. doi: 10.1093/brain/awm135. - DOI - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
