

Statistical potential for modeling and ranking of protein-ligand interactions
- PMID: 22014038
- PMCID: PMC3246566
- DOI: 10.1021/ci200377u
Statistical potential for modeling and ranking of protein-ligand interactions
Abstract
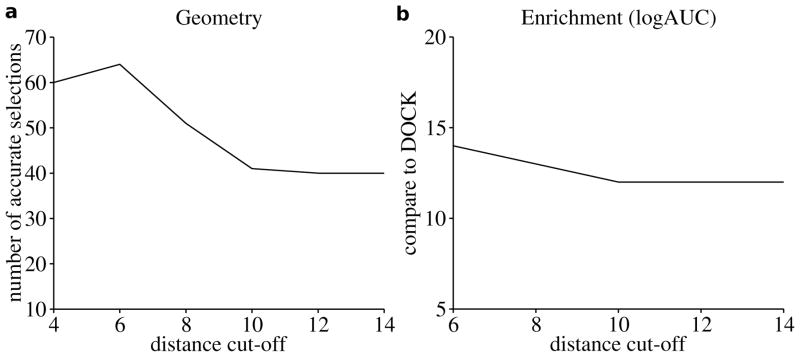
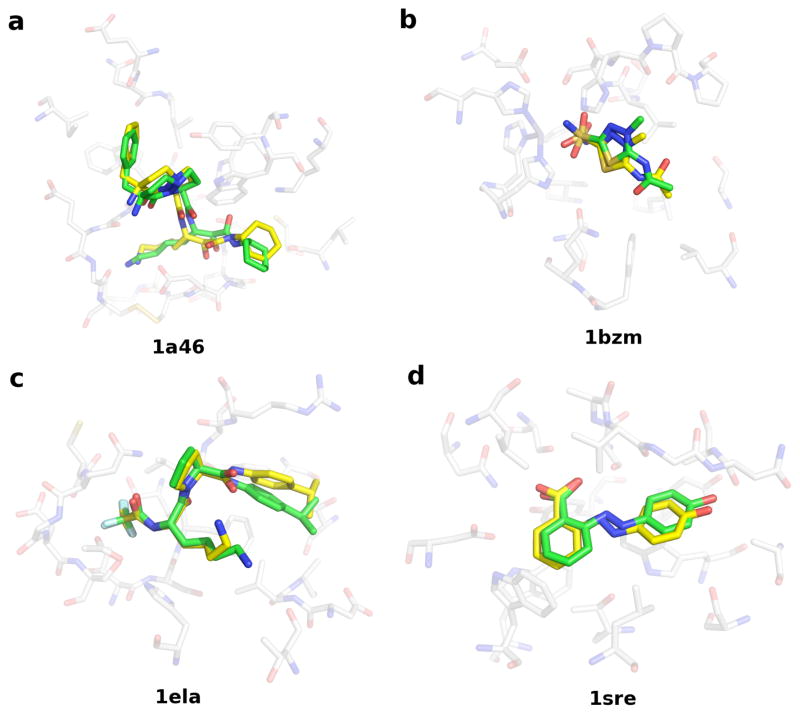
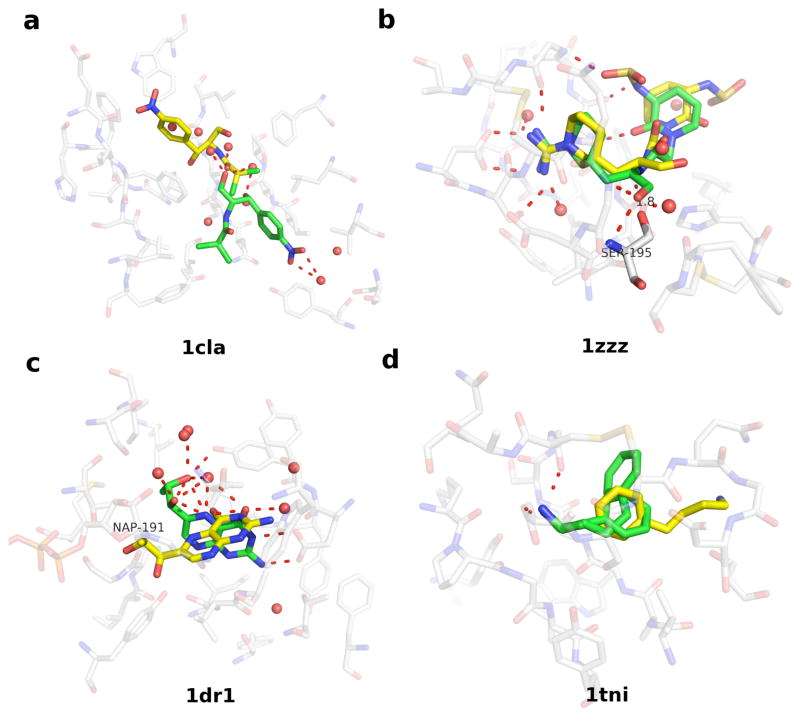
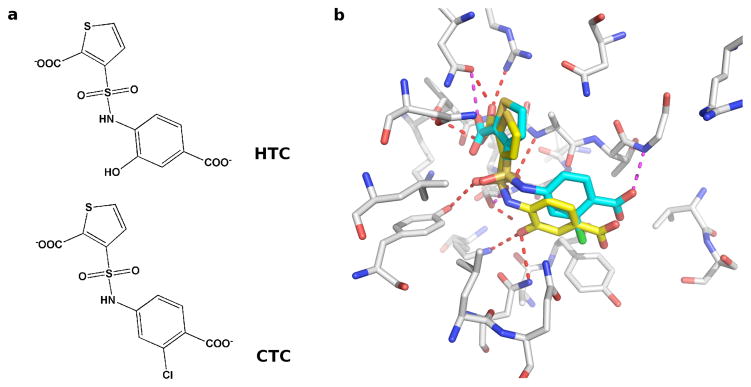
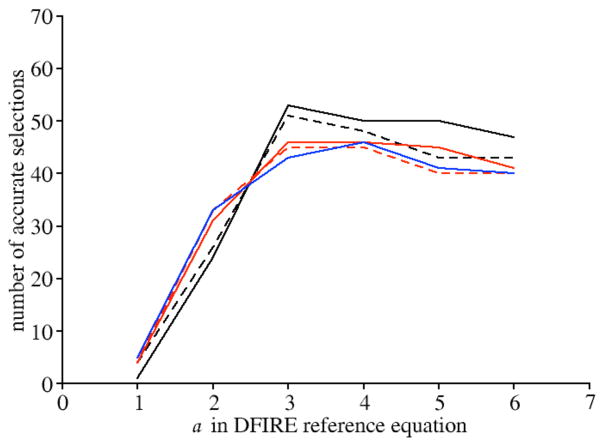
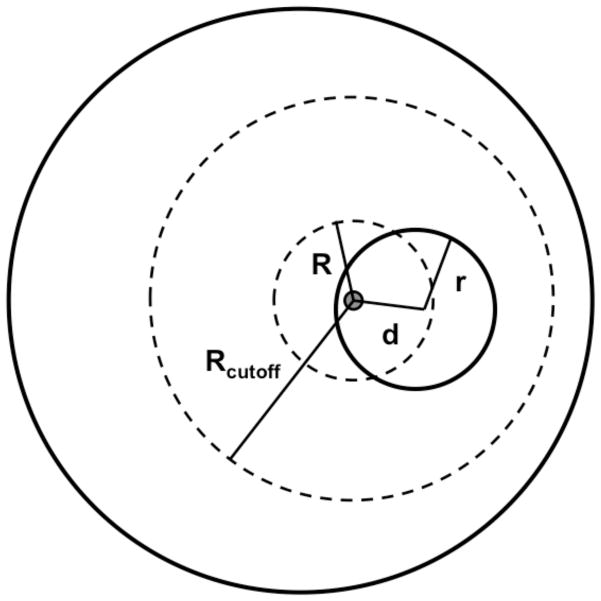
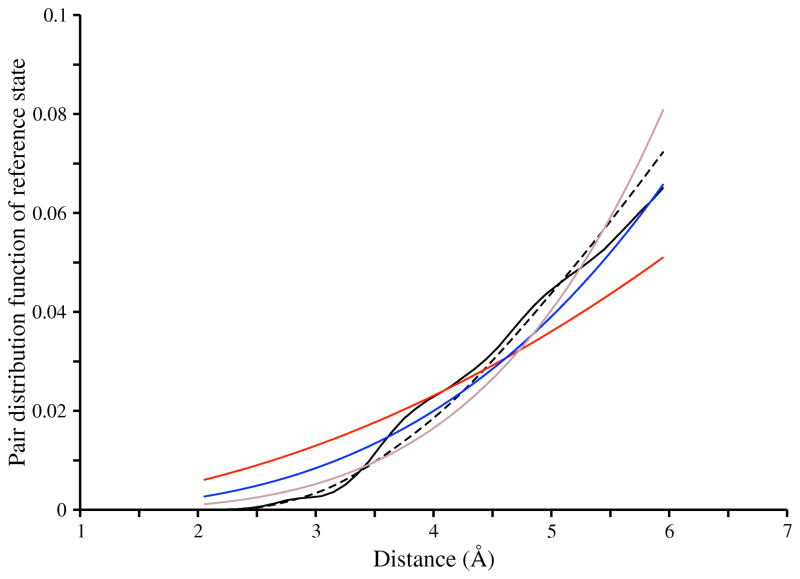
Applications in structural biology and medicinal chemistry require protein-ligand scoring functions for two distinct tasks: (i) ranking different poses of a small molecule in a protein binding site and (ii) ranking different small molecules by their complementarity to a protein site. Using probability theory, we developed two atomic distance-dependent statistical scoring functions: PoseScore was optimized for recognizing native binding geometries of ligands from other poses and RankScore was optimized for distinguishing ligands from nonbinding molecules. Both scores are based on a set of 8,885 crystallographic structures of protein-ligand complexes but differ in the values of three key parameters. Factors influencing the accuracy of scoring were investigated, including the maximal atomic distance and non-native ligand geometries used for scoring, as well as the use of protein models instead of crystallographic structures for training and testing the scoring function. For the test set of 19 targets, RankScore improved the ligand enrichment (logAUC) and early enrichment (EF(1)) scores computed by DOCK 3.6 for 13 and 14 targets, respectively. In addition, RankScore performed better at rescoring than each of seven other scoring functions tested. Accepting both the crystal structure and decoy geometries with all-atom root-mean-square errors of up to 2 Å from the crystal structure as correct binding poses, PoseScore gave the best score to a correct binding pose among 100 decoys for 88% of all cases in a benchmark set containing 100 protein-ligand complexes. PoseScore accuracy is comparable to that of DrugScore(CSD) and ITScore/SE and superior to 12 other tested scoring functions. Therefore, RankScore can facilitate ligand discovery, by ranking complexes of the target with different small molecules; PoseScore can be used for protein-ligand complex structure prediction, by ranking different conformations of a given protein-ligand pair. The statistical potentials are available through the Integrative Modeling Platform (IMP) software package (http://salilab.org/imp) and the LigScore Web server (http://salilab.org/ligscore/).
Figures
Similar articles
-
DrugScore(CSD)-knowledge-based scoring function derived from small molecule crystal data with superior recognition rate of near-native ligand poses and better affinity prediction.J Med Chem. 2005 Oct 6;48(20):6296-303. doi: 10.1021/jm050436v. J Med Chem. 2005. PMID: 16190756
-
Boosted neural networks scoring functions for accurate ligand docking and ranking.J Bioinform Comput Biol. 2018 Apr;16(2):1850004. doi: 10.1142/S021972001850004X. Epub 2018 Feb 4. J Bioinform Comput Biol. 2018. PMID: 29495922
-
A knowledge-based energy function for protein-ligand, protein-protein, and protein-DNA complexes.J Med Chem. 2005 Apr 7;48(7):2325-35. doi: 10.1021/jm049314d. J Med Chem. 2005. PMID: 15801826
-
Scoring functions for prediction of protein-ligand interactions.Curr Pharm Des. 2013;19(12):2174-82. doi: 10.2174/1381612811319120005. Curr Pharm Des. 2013. PMID: 23016847 Review.
-
Advances in Docking.Curr Med Chem. 2019;26(42):7555-7580. doi: 10.2174/0929867325666180904115000. Curr Med Chem. 2019. PMID: 30182836 Review.
Cited by
-
Aromatic interactions at the ligand-protein interface: Implications for the development of docking scoring functions.Chem Biol Drug Des. 2018 Feb;91(2):380-390. doi: 10.1111/cbdd.13084. Epub 2017 Aug 31. Chem Biol Drug Des. 2018. PMID: 28816025 Free PMC article.
-
Archiving and disseminating integrative structure models.J Biomol NMR. 2019 Jul;73(6-7):385-398. doi: 10.1007/s10858-019-00264-2. Epub 2019 Jul 5. J Biomol NMR. 2019. PMID: 31278630 Free PMC article.
-
Optimized atomic statistical potentials: assessment of protein interfaces and loops.Bioinformatics. 2013 Dec 15;29(24):3158-66. doi: 10.1093/bioinformatics/btt560. Epub 2013 Sep 27. Bioinformatics. 2013. PMID: 24078704 Free PMC article.
-
Molecular modeling and ligand docking for solute carrier (SLC) transporters.Curr Top Med Chem. 2013;13(7):843-56. doi: 10.2174/1568026611313070007. Curr Top Med Chem. 2013. PMID: 23578028 Free PMC article. Review.
-
Extracting knowledge from protein structure geometry.Proteins. 2013 May;81(5):841-51. doi: 10.1002/prot.24242. Epub 2013 Feb 27. Proteins. 2013. PMID: 23280479 Free PMC article.
References
-
- Brooijmans N, Kuntz ID. Molecular recognition and docking algorithms. Annu Rev Biophys Biomol Struct. 2003;32:335–373. - PubMed
-
- Leach AR, Shoichet BK, Peishoff CE. Prediction of protein-ligand interactions. Docking and scoring: Successes and gaps. J Med Chem. 2006;49(20):5851–5855. - PubMed
-
- Gilson MK, Zhou HX. Calculation of protein-ligand binding affinities. Annu Rev Biophys Biomol Struct. 2007:3621–42. - PubMed
-
- Jain AN. Scoring functions for protein-ligand docking. Curr Protein Pept Sci. 2006;7(5):407–420. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
