

Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms
- PMID: 22014092
- PMCID: PMC3225693
- DOI: 10.1111/j.1365-2958.2011.07877.x
Quantitative proteomics reveals metabolic and pathogenic properties of Chlamydia trachomatis developmental forms
Abstract
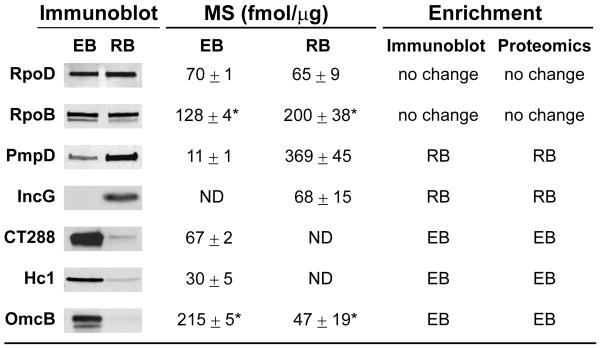
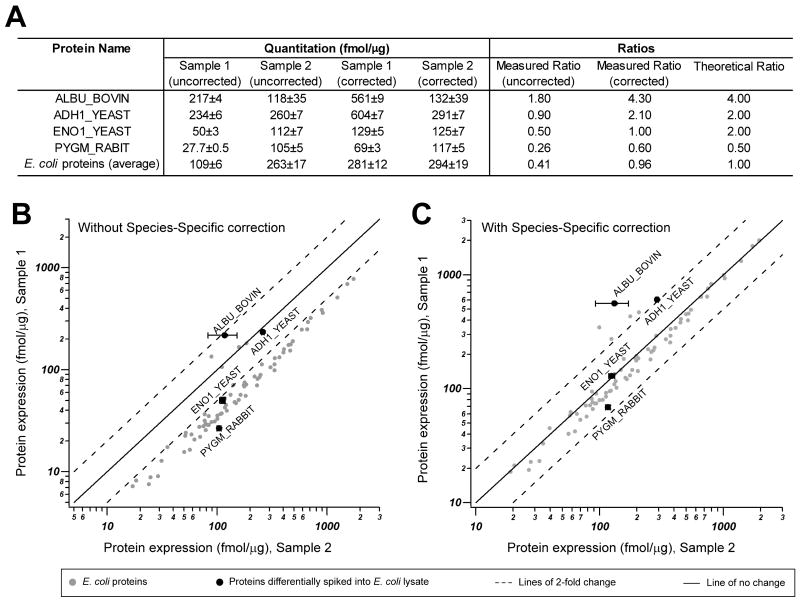
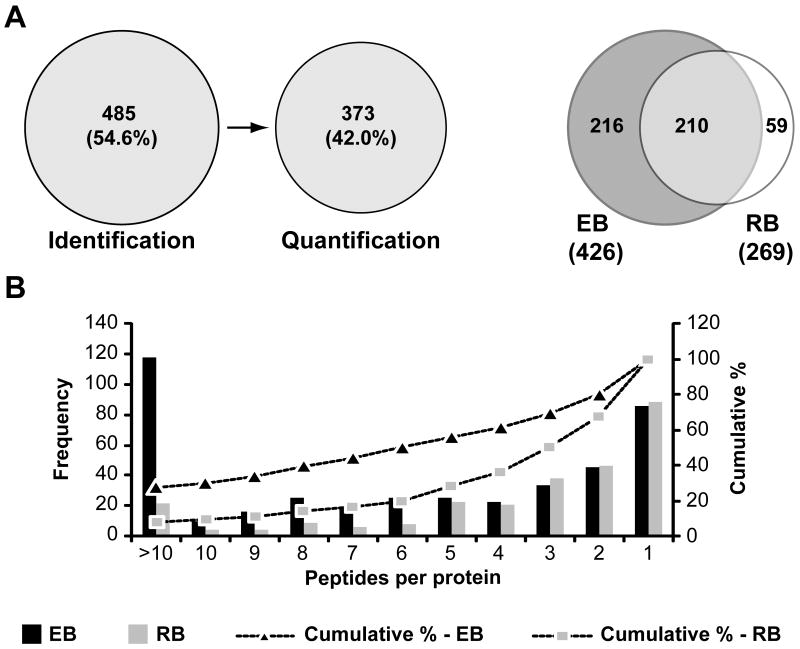
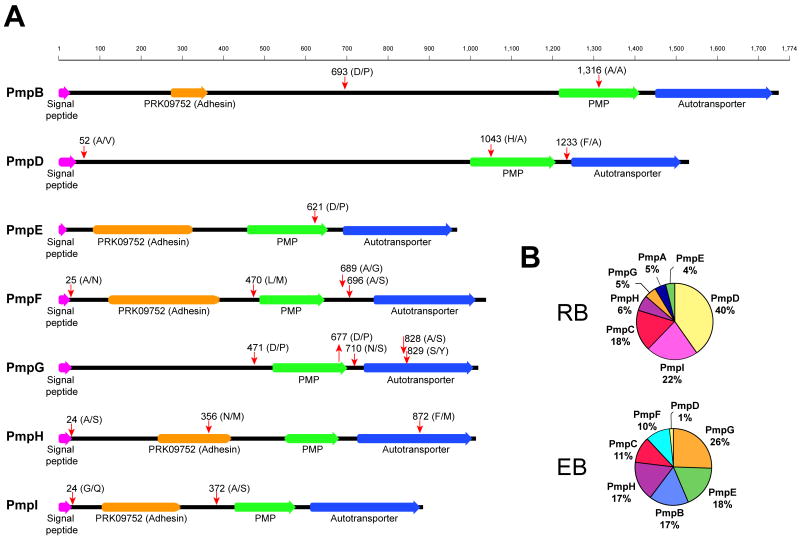
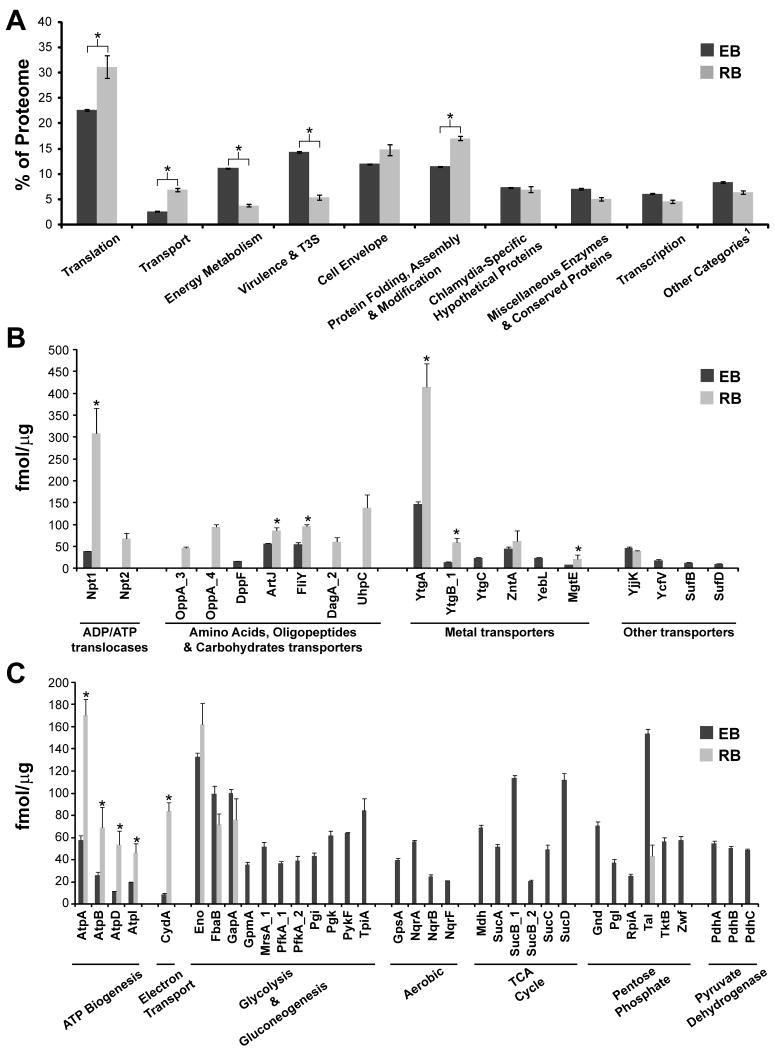
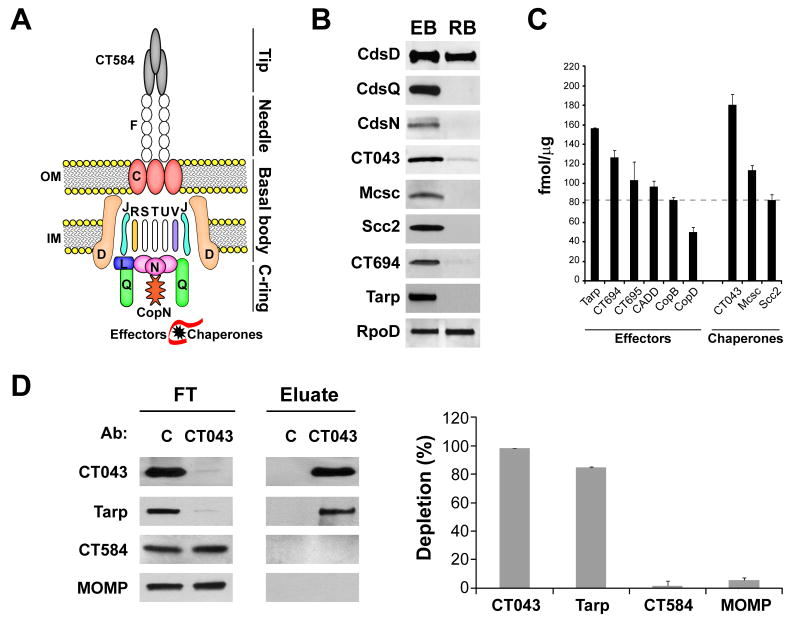
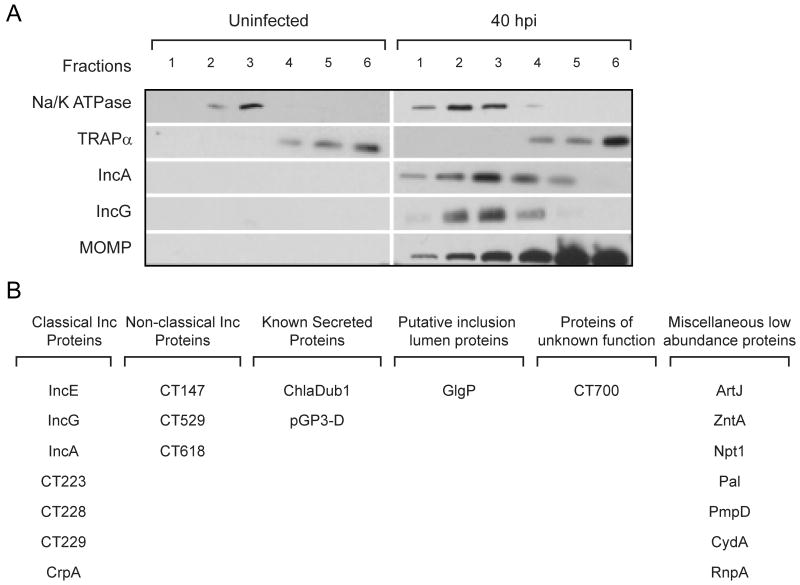
Chlamydia trachomatis is an obligate intracellular pathogen responsible for ocular and genital infections of significant public health importance. C. trachomatis undergoes a biphasic developmental cycle alternating between two distinct forms: the infectious elementary body (EB), and the replicative but non-infectious reticulate body (RB). The molecular basis for these developmental transitions and the metabolic properties of the EB and RB forms are poorly understood as these bacteria have traditionally been difficult to manipulate through classical genetic approaches. Using two-dimensional liquid chromatography - tandem mass spectrometry (LC/LC-MS/MS) we performed a large-scale, label-free quantitative proteomic analysis of C. trachomatis LGV-L2 EB and RB forms. Additionally, we carried out LC-MS/MS to analyse the membranes of the pathogen-containing vacuole ('inclusion'). We developed a label-free quantification approaches to measure protein abundance in a mixed-proteome background which we applied for EB and RB quantitative analysis. In this manner, we catalogued the relative distribution of > 54% of the predicted proteins in the C. trachomatis LGV-L2 proteome. Proteins required for central metabolism and glucose catabolism were predominant in the EB, whereas proteins associated with protein synthesis, ATP generation and nutrient transport were more abundant in the RB. These findings suggest that the EB is primed for a burst in metabolic activity upon entry, whereas the RB form is geared towards nutrient utilization, a rapid increase in cellular mass, and securing the resources for an impending transition back to the EB form. The most revealing difference between the two forms was the relative deficiency of cytoplasmic factors required for efficient type III secretion (T3S) in the RB stage at 18 h post infection, suggesting a reduced T3S capacity or a low frequency of active T3S apparatus assembled on a 'per organism' basis. Our results show that EB and RB proteomes are streamlined to fulfil their predicted biological functions: maximum infectivity for EBs and replicative capacity for RBs.
© 2011 Blackwell Publishing Ltd.
Figures
References
-
- Bannantine JP, Griffiths RS, Viratyosin W, Brown WJ, Rockey DD. A secondary structure motif predictive of protein localization to the chlamydial inclusion membrane. Cell Microbiol. 2000;2:35–47. - PubMed
-
- Bebear C, de Barbeyrac B. Genital Chlamydia trachomatis infections. Clin Microbiol Infect. 2009;15:4–10. - PubMed
-
- Beeckman DS, Vanrompay DC. Bacterial secretion systems with an emphasis on the chlamydial Type III secretion system. Curr Issues Mol Biol. 2010;12:17–41. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
