

Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: phylogeny, sequence variability, and SNP distribution
- PMID: 22016062
- PMCID: PMC3231845
- DOI: 10.1167/iovs.11-7812
Multiplex sequencing of seven ocular herpes simplex virus type-1 genomes: phylogeny, sequence variability, and SNP distribution
Abstract
Purpose: Little is known about the role of sequence variation in the pathology of HSV-1 keratitis virus. The goal was to show that a multiplex, high-throughput genome-sequencing approach is feasible for simultaneously sequencing seven HSV-1 ocular strains.
Methods: A genome sequencer was used to sequence the HSV-1 ocular isolates TFT401, 134, CJ311, CJ360, CJ394, CJ970, and OD4, in a single lane. Reads were mapped to the HSV-1 strain 17 reference genome by high-speed sequencing. ClustalW was used for alignment, and the Mega 4 package was used for phylogenetic analysis (www.megasoftware.net). Simplot was used to compare genetic variability and high-speed sequencing was used to identify SNPs (developed by Stuart Ray, Johns Hopkins University School of Medicine, Baltimore, MD, http://sray.med.som.jhml.edu/SCRoftware/simplot).
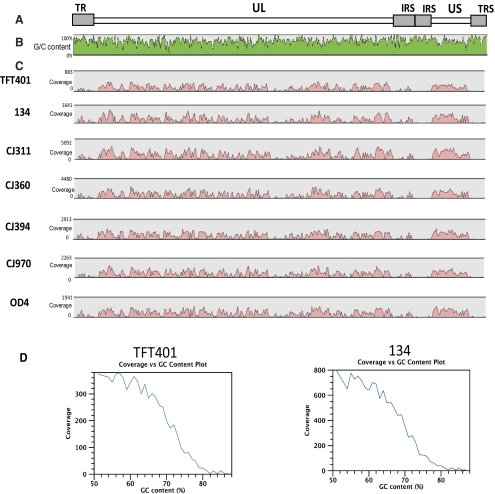
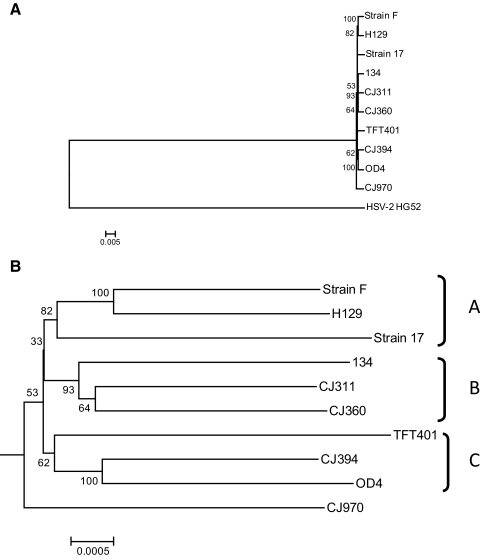
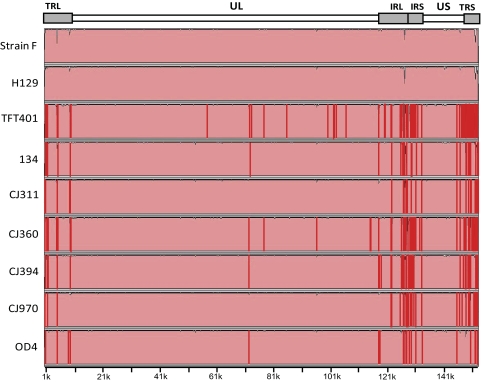
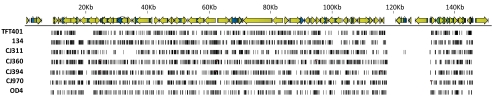
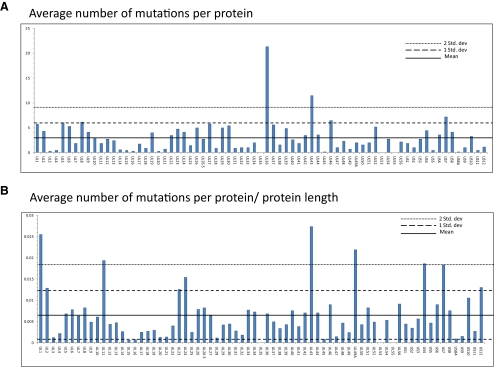
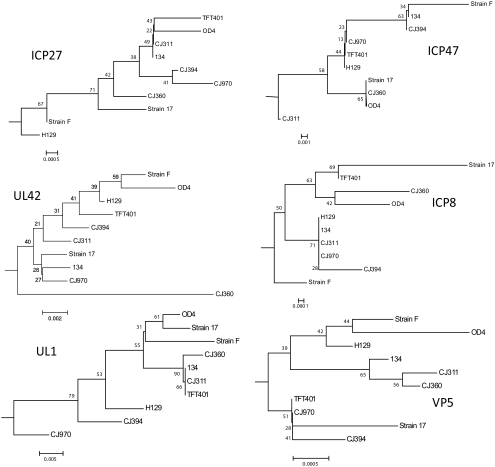
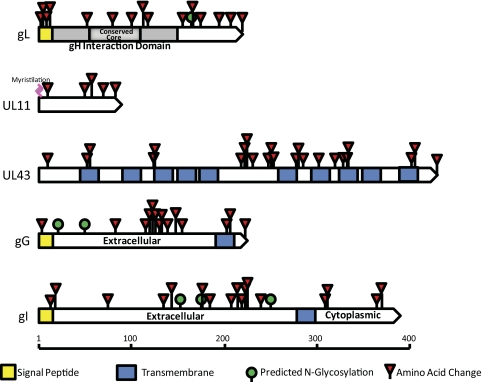
Results: Approximately 95% to 99% of the seven genomes were sequenced in a single lane with average coverage ranging from 224 to 1345. Phylogenetic analysis of the sequenced genome regions revealed at least three clades. Each strain had approximately 200 coding SNPs compared to strain 17, and these were evenly spaced along the genomes. Four genes were highly conserved, and six were more variable. Reduced coverage was obtained in the highly GC-rich terminal repeat regions.
Conclusions: Multiplex sequencing is a cost-effective way to obtain the genomic sequences of ocular HSV-1 isolates with sufficient coverage of the unique regions for genomic analysis. The number of SNPs and their distribution will be useful for analyzing the genetics of virulence, and the sequence data will be useful for studying HSV-1 evolution and for the design of structure-function studies.
Figures

Similar articles
-
Sequence variation in the herpes simplex virus U(S)1 ocular virulence determinant.Invest Ophthalmol Vis Sci. 2011 Jun 28;52(7):4630-8. doi: 10.1167/iovs.10-7032. Invest Ophthalmol Vis Sci. 2011. PMID: 21519032 Free PMC article.
-
Polymorphisms of thymidine kinase gene in herpes simplex virus type 1: analysis of clinical isolates from herpetic keratitis patients and laboratory strains.J Med Virol. 1998 Oct;56(2):151-8. doi: 10.1002/(sici)1096-9071(199810)56:2<151::aid-jmv9>3.0.co;2-7. J Med Virol. 1998. PMID: 9746072
-
Multiple determinants contribute to the virulence of HSV ocular and CNS infection and identification of serine 34 of the US1 gene as an ocular disease determinant.Invest Ophthalmol Vis Sci. 2003 Jun;44(6):2657-68. doi: 10.1167/iovs.02-1105. Invest Ophthalmol Vis Sci. 2003. PMID: 12766070
-
Comparison of Herpes Simplex Virus 1 Strains Circulating in Finland Demonstrates the Uncoupling of Whole-Genome Relatedness and Phenotypic Outcomes of Viral Infection.J Virol. 2019 Apr 3;93(8):e01824-18. doi: 10.1128/JVI.01824-18. Print 2019 Apr 15. J Virol. 2019. PMID: 30760568 Free PMC article.
-
History and genomic sequence analysis of the herpes simplex virus 1 KOS and KOS1.1 sub-strains.Virology. 2016 Jan;487:215-21. doi: 10.1016/j.virol.2015.09.026. Epub 2015 Nov 5. Virology. 2016. PMID: 26547038 Free PMC article. Review.
Cited by
-
Global Diversity within and between Human Herpesvirus 1 and 2 Glycoproteins.J Virol. 2015 Aug;89(16):8206-18. doi: 10.1128/JVI.01302-15. Epub 2015 May 27. J Virol. 2015. PMID: 26018161 Free PMC article.
-
Quantitative Trait Locus Based Virulence Determinant Mapping of the HSV-1 Genome in Murine Ocular Infection: Genes Involved in Viral Regulatory and Innate Immune Networks Contribute to Virulence.PLoS Pathog. 2016 Mar 10;12(3):e1005499. doi: 10.1371/journal.ppat.1005499. eCollection 2016 Mar. PLoS Pathog. 2016. PMID: 26962864 Free PMC article.
-
Surveillance for feline herpesvirus type 1 mutation and development of resistance in cats treated with antiviral medications.Front Vet Sci. 2023 May 18;10:1197249. doi: 10.3389/fvets.2023.1197249. eCollection 2023. Front Vet Sci. 2023. PMID: 37275610 Free PMC article.
-
Evolution and diversity in human herpes simplex virus genomes.J Virol. 2014 Jan;88(2):1209-27. doi: 10.1128/JVI.01987-13. Epub 2013 Nov 13. J Virol. 2014. PMID: 24227835 Free PMC article.
-
Genotypic Characterization of Herpes Simplex Virus Type 1 Isolates in Immunocompromised Patients in Rio de Janeiro, Brazil.PLoS One. 2015 Sep 25;10(9):e0136825. doi: 10.1371/journal.pone.0136825. eCollection 2015. PLoS One. 2015. PMID: 26407292 Free PMC article. Clinical Trial.
References
-
- Liesegang TJ. Herpes simplex virus epidemiology and ocular importance. Cornea. 2001;20:1–13 - PubMed
-
- Whitley RJ. Herpes simplex viruses. In: Fields BN, Knipe DM, Howley PM, eds. Fields Virology. 3rd ed Vol. 2 Philadelphia: Lippincott-Raven; 1996:2297–2342
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
