

Direct mutation analysis by high-throughput sequencing: from germline to low-abundant, somatic variants
- PMID: 22016070
- PMCID: PMC3237897
- DOI: 10.1016/j.mrfmmm.2011.10.001
Direct mutation analysis by high-throughput sequencing: from germline to low-abundant, somatic variants
Abstract
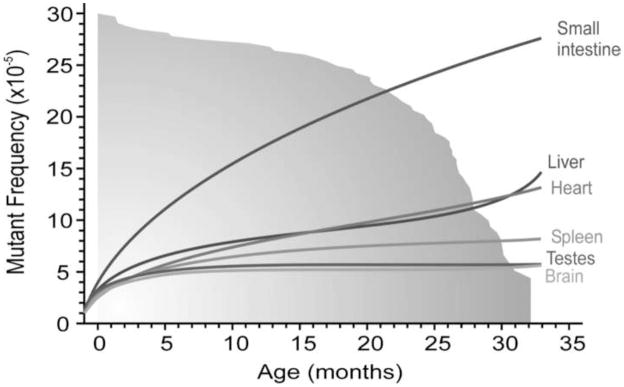
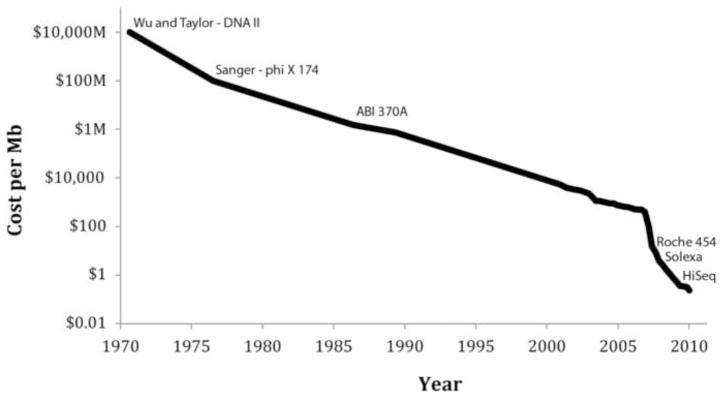
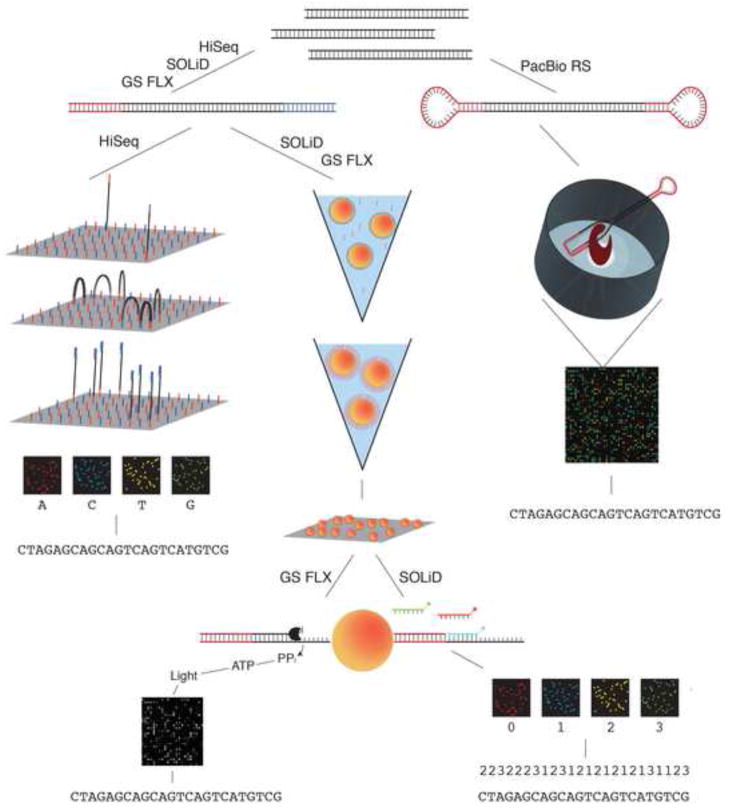
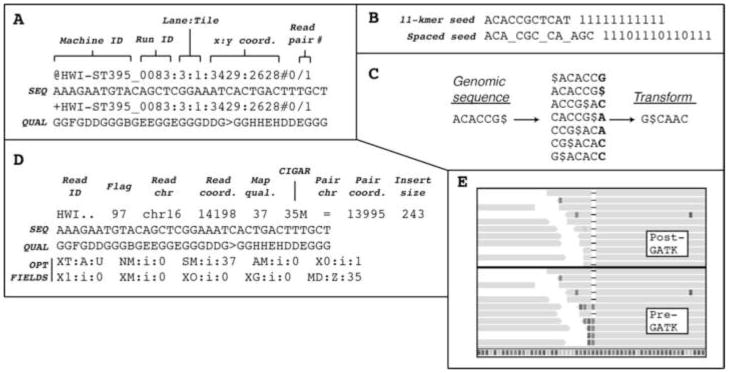
DNA mutations are the source of genetic variation within populations. The majority of mutations with observable effects are deleterious. In humans mutations in the germ line can cause genetic disease. In somatic cells multiple rounds of mutations and selection lead to cancer. The study of genetic variation has progressed rapidly since the completion of the draft sequence of the human genome. Recent advances in sequencing technology, most importantly the introduction of massively parallel sequencing (MPS), have resulted in more than a hundred-fold reduction in the time and cost required for sequencing nucleic acids. These improvements have greatly expanded the use of sequencing as a practical tool for mutation analysis. While in the past the high cost of sequencing limited mutation analysis to selectable markers or small forward mutation targets assumed to be representative for the genome overall, current platforms allow whole genome sequencing for less than $5000. This has already given rise to direct estimates of germline mutation rates in multiple organisms including humans by comparing whole genome sequences between parents and offspring. Here we present a brief history of the field of mutation research, with a focus on classical tools for the measurement of mutation rates. We then review MPS, how it is currently applied and the new insight into human and animal mutation frequencies and spectra that has been obtained from whole genome sequencing. While great progress has been made, we note that the single most important limitation of current MPS approaches for mutation analysis is the inability to address low-abundance mutations that turn somatic tissues into mosaics of cells. Such mutations are at the basis of intra-tumor heterogeneity, with important implications for clinical diagnosis, and could also contribute to somatic diseases other than cancer, including aging. Some possible approaches to gain access to low-abundance mutations are discussed, with a brief overview of new sequencing platforms that are currently waiting in the wings to advance this exploding field even further.
Copyright © 2011 Elsevier B.V. All rights reserved.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Figures
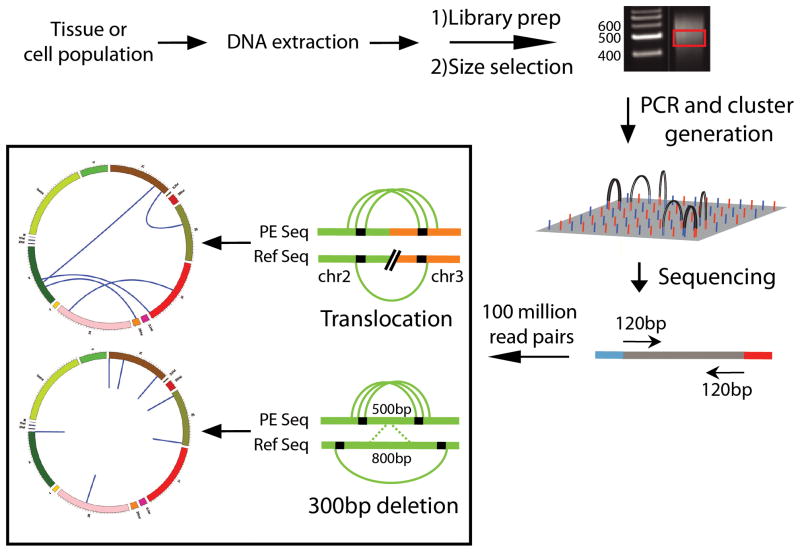
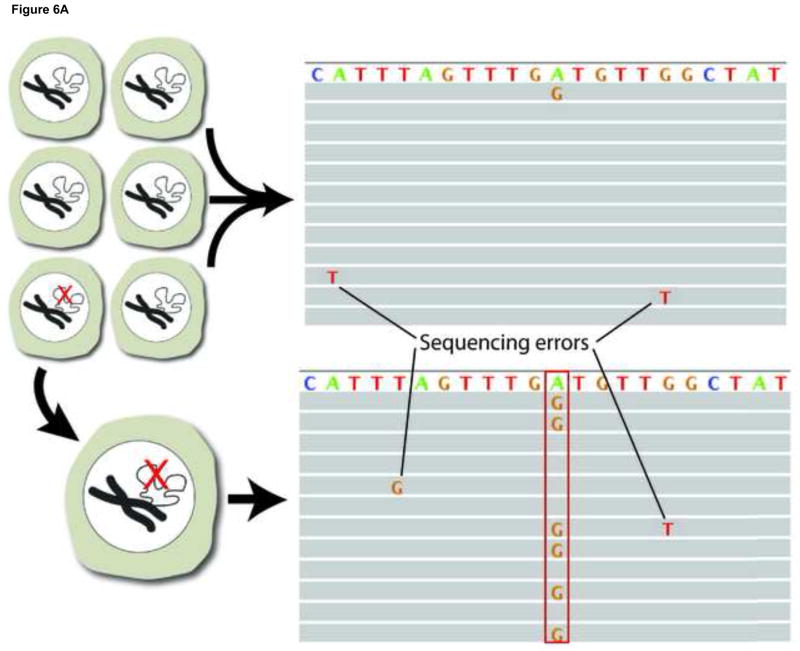
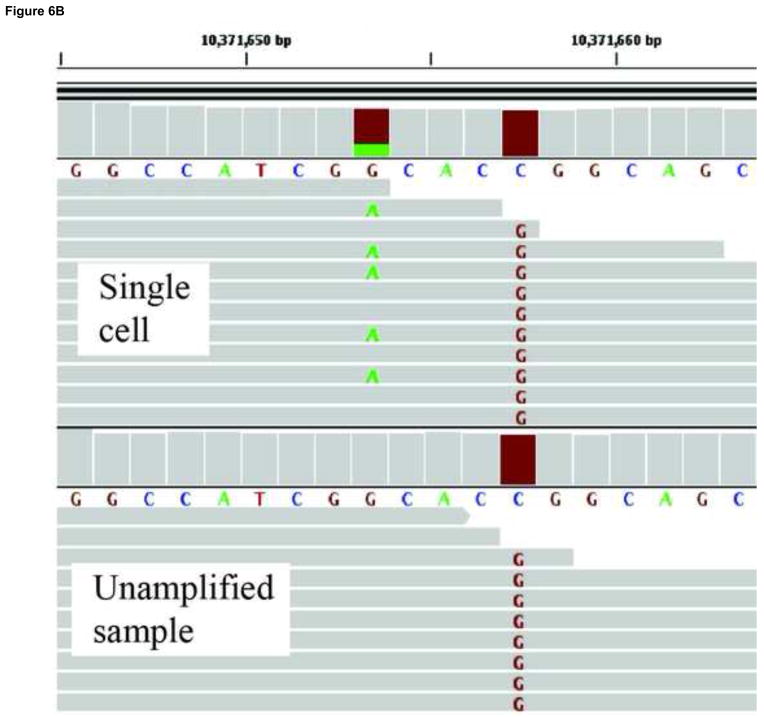
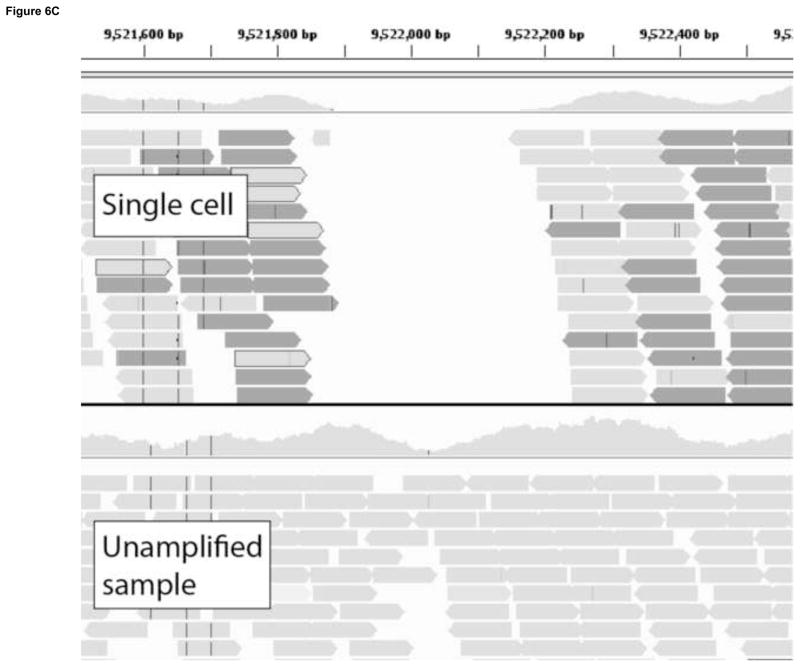
References
-
- Castle WE. The Mutation Theory of Organic Evolution, from the Standpoint of Animal Breeding. Science. 1905;21:521–525. - PubMed
-
- Muller HJ. Further changes in the white-eye series of Drosophila and their bearing on the manner of occurrence of mutation. Journal of Experimental Zoology. 1920;31:443–474.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
