

Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1
- PMID: 22016541
- PMCID: PMC3202339
- DOI: 10.1523/JNEUROSCI.2443-11.2011
Resolving TRPV1- and TNF-α-mediated spinal cord synaptic plasticity and inflammatory pain with neuroprotectin D1
Abstract
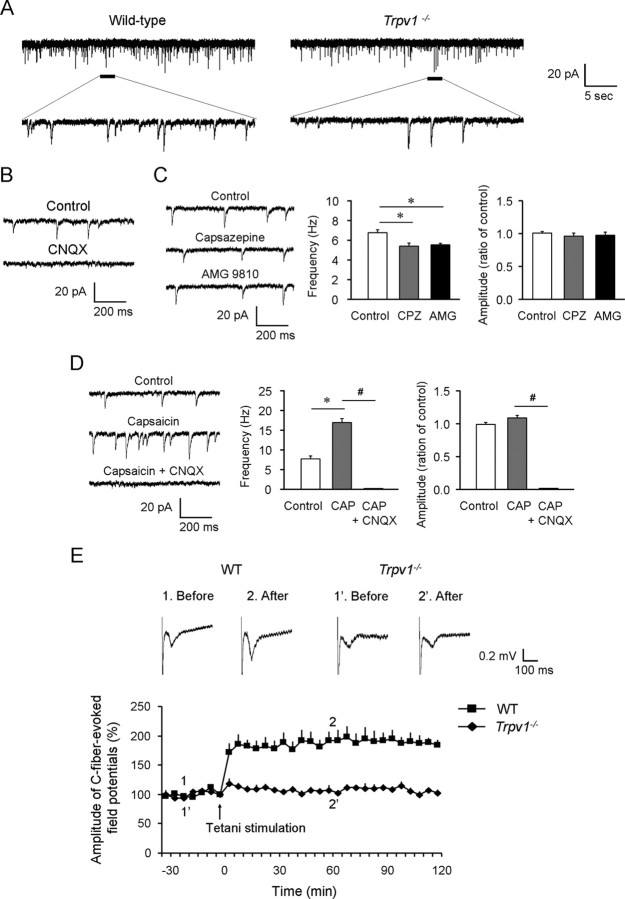
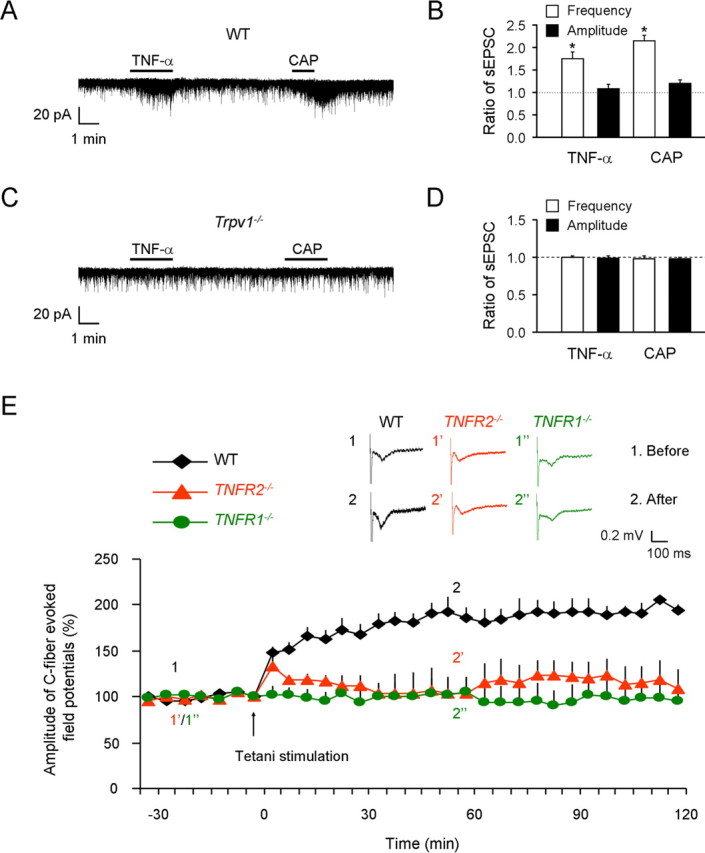
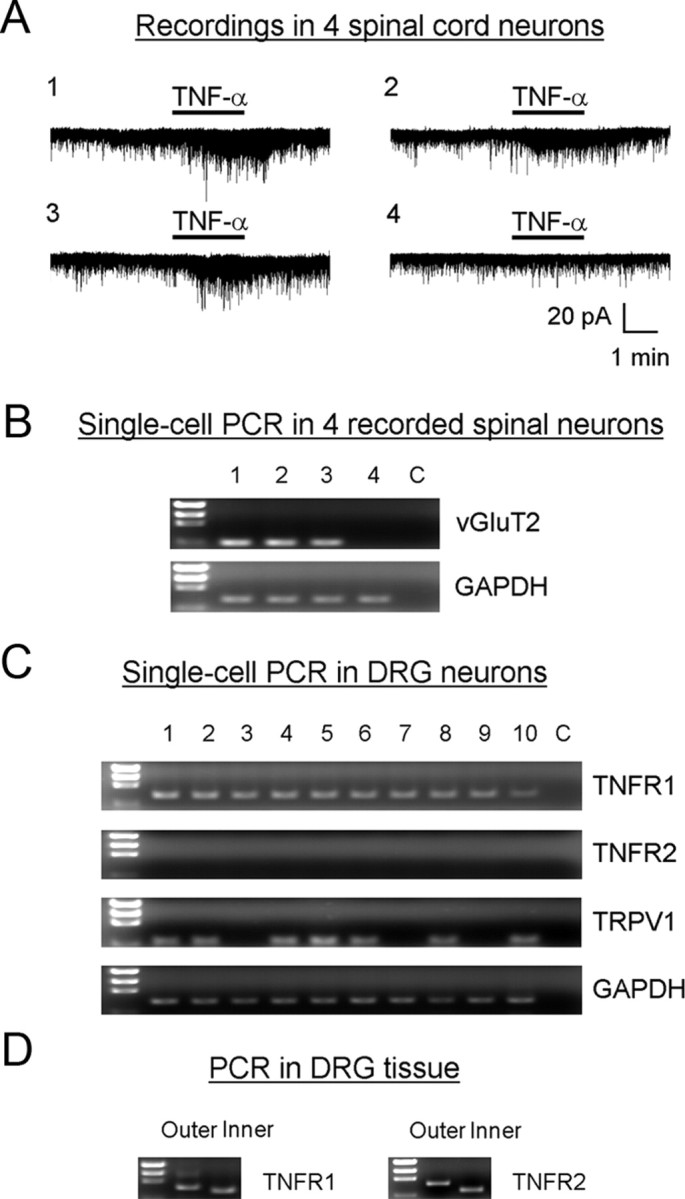
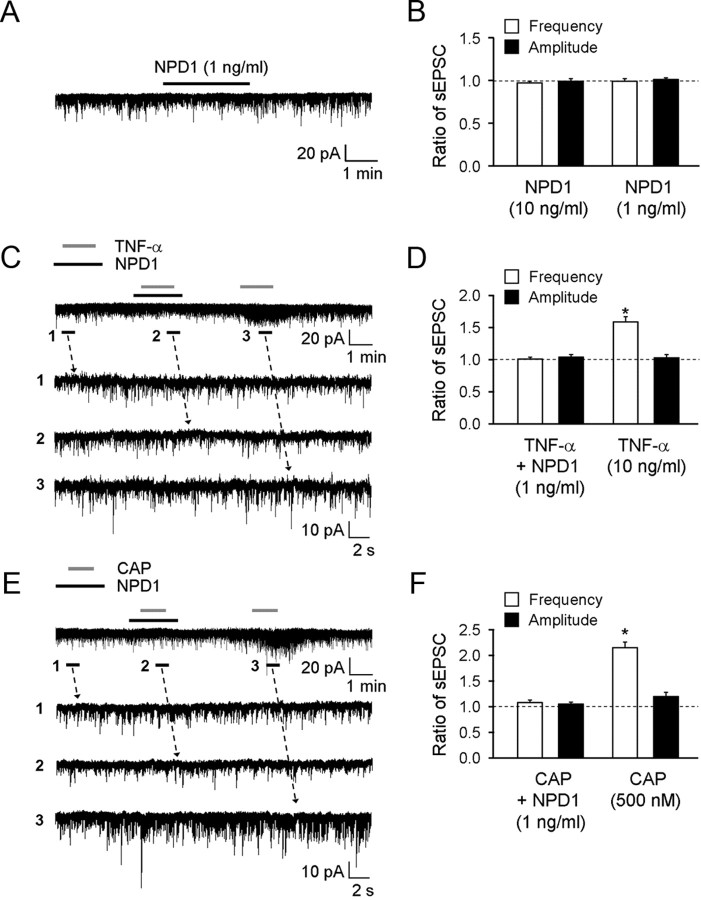
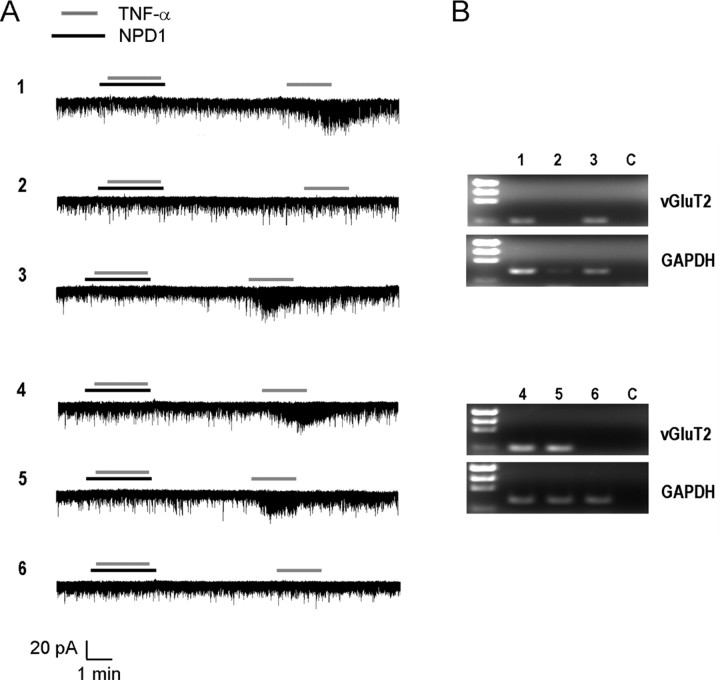
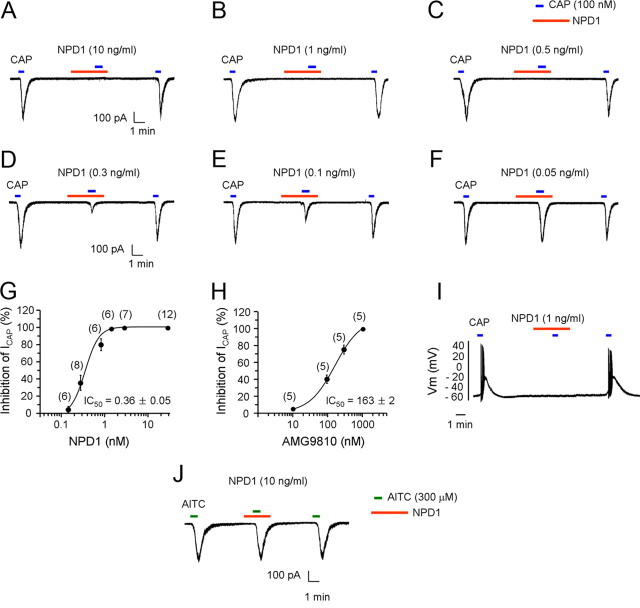
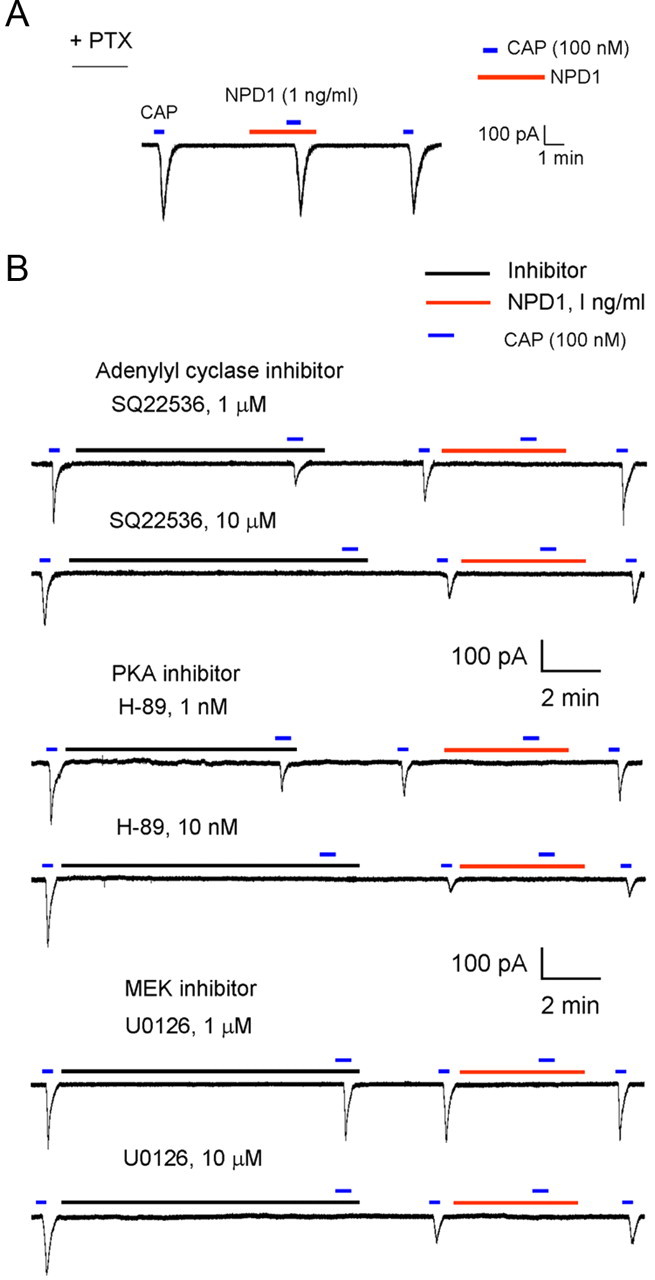
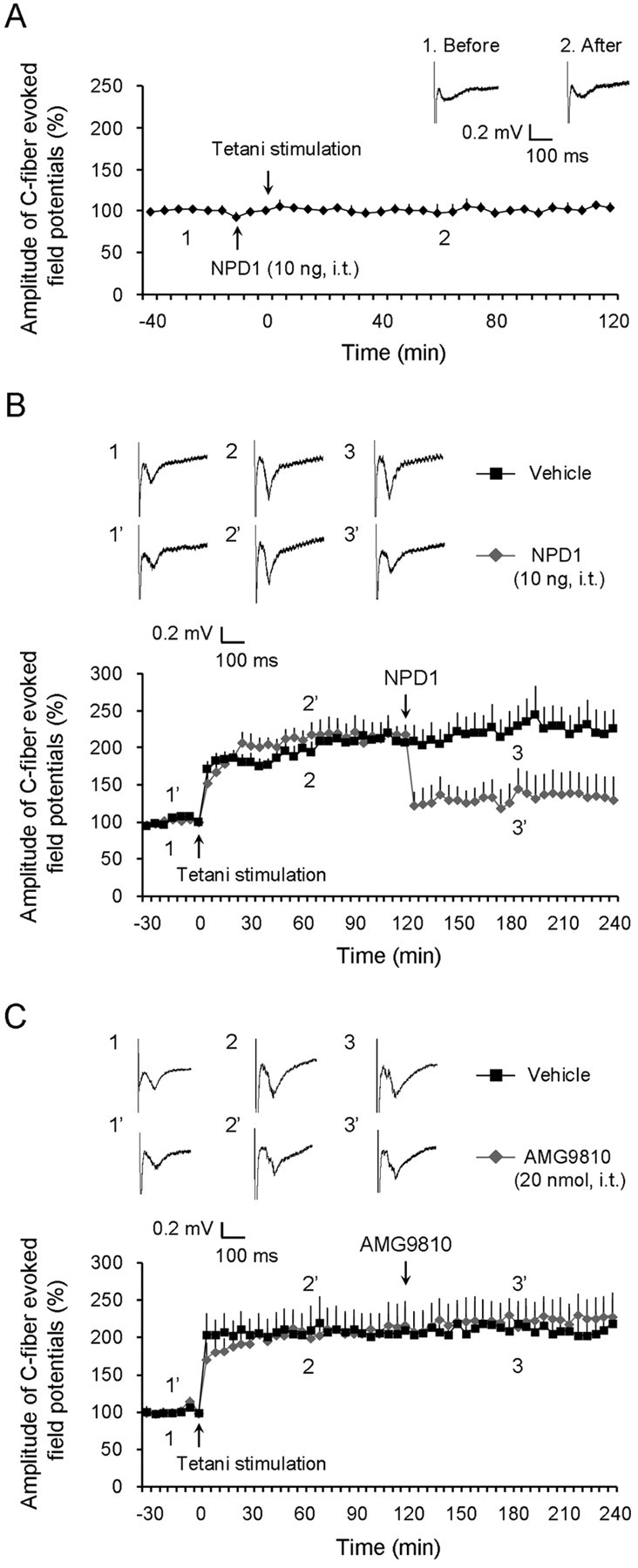
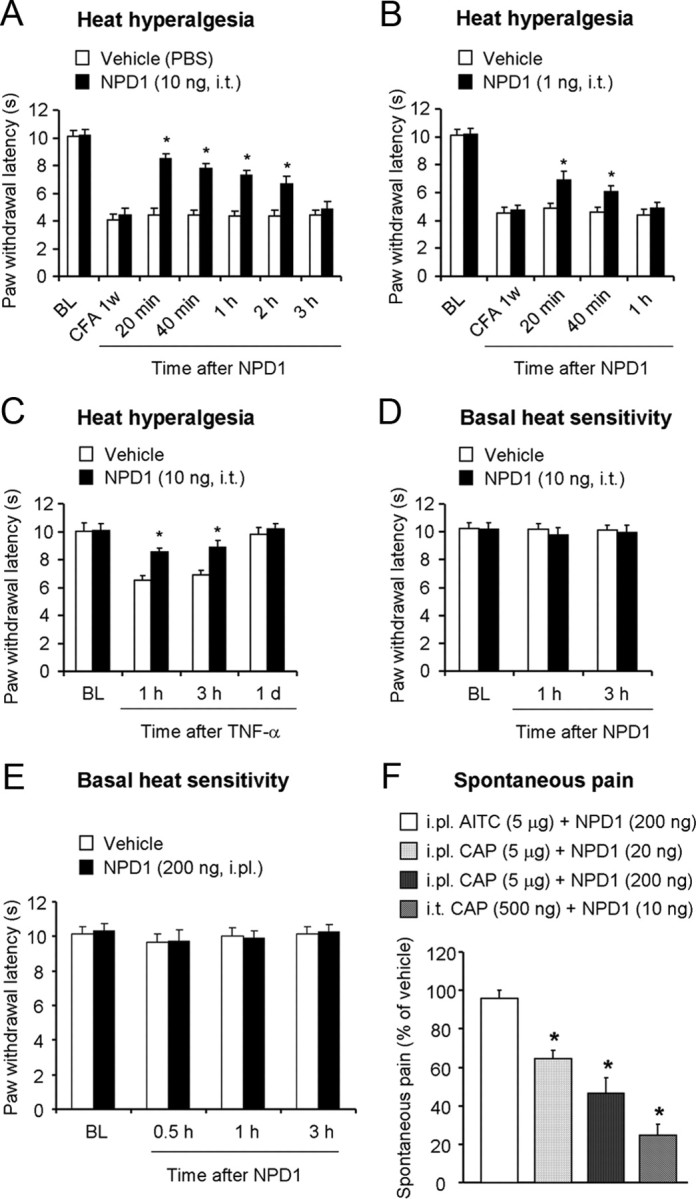
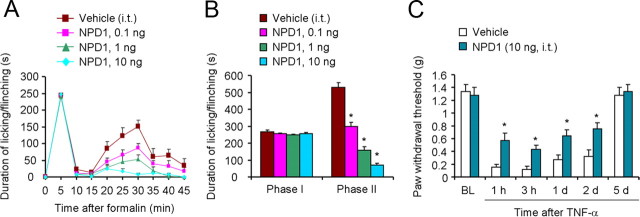
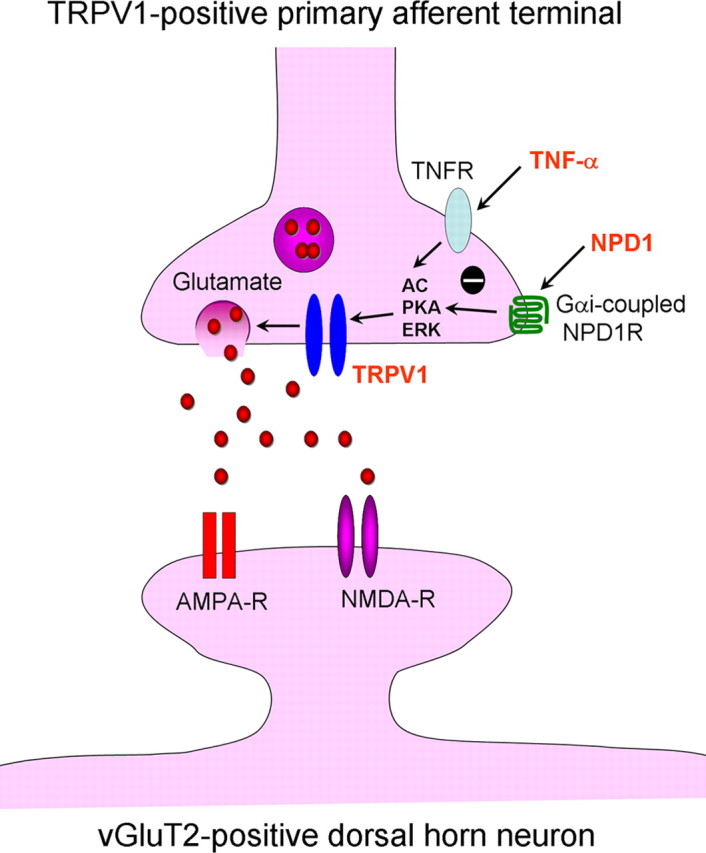
Mechanisms of inflammatory pain are not fully understood. We investigated the role of TRPV1 (transient receptor potential subtype V1) and TNF-α, two critical mediators for inflammatory pain, in regulating spinal cord synaptic transmission. We found in mice lacking Trpv1 the frequency but not the amplitude of spontaneous EPSCs (sEPSCs) in lamina II neurons of spinal cord slices is reduced. Further, C-fiber-induced spinal long-term potentiation (LTP) in vivo is abolished in Trpv1 knock-out mice. TNF-α also increases sEPSC frequency but not amplitude in spinal outer lamina II (lamina IIo) neurons, and this increase is abolished in Trpv1 knock-out mice. Single-cell PCR analysis revealed that TNF-α-responding neurons in lamina IIo are exclusively excitatory (vGluT2(+)) neurons. Notably, neuroprotectin-1 (NPD1), an anti-inflammatory lipid mediator derived from ω-3 polyunsaturated fatty acid (docosahexaenoic acid), blocks TNF-α- and capsaicin-evoked sEPSC frequency increases but has no effect on basal synaptic transmission. Strikingly, NPD1 potently inhibits capsaicin-induced TRPV1 current (IC(50) = 0.4 nm) in dissociated dorsal root ganglion neurons, and this IC(50) is ≈ 500 times lower than that of AMG9810, a commonly used TRPV1 antagonist. NPD1 inhibition of TRPV1 is mediated by GPCRs, since the effects were blocked by pertussis toxin. In contrast, NPD1 had no effect on mustard oil-induced TRPA1 currents. Spinal injection of NPD1, at very low doses (0.1-10 ng), blocks spinal LTP and reduces TRPV1-dependent inflammatory pain, without affecting baseline pain. NPD1 also reduces TRPV1-independent but TNF-α-dependent pain hypersensitivity. Our findings demonstrate a novel role of NPD1 in regulating TRPV1/TNF-α-mediated spinal synaptic plasticity and identify NPD1 as a novel analgesic for treating inflammatory pain.
Figures
References
-
- Beattie EC, Stellwagen D, Morishita W, Bresnahan JC, Ha BK, Von Zastrow M, Beattie MS, Malenka RC. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–2285. - PubMed
-
- Bhave G, Zhu W, Wang H, Brasier DJ, Oxford GS, Gereau RW., 4th cAMP-dependent protein kinase regulates desensitization of the capsaicin receptor (VR1) by direct phosphorylation. Neuron. 2002;35:721–731. - PubMed
-
- Bito H, Nakamura M, Honda Z, Izumi T, Iwatsubo T, Seyama Y, Ogura A, Kudo Y, Shimizu T. Platelet-activating factor (PAF) receptor in rat brain: PAF mobilizes intracellular Ca2+ in hippocampal neurons. Neuron. 1992;9:285–294. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases