

HIV-1gp120 induces neuronal apoptosis through enhancement of 4-aminopyridine-senstive outward K+ currents
- PMID: 22016798
- PMCID: PMC3189248
- DOI: 10.1371/journal.pone.0025994
HIV-1gp120 induces neuronal apoptosis through enhancement of 4-aminopyridine-senstive outward K+ currents
Abstract
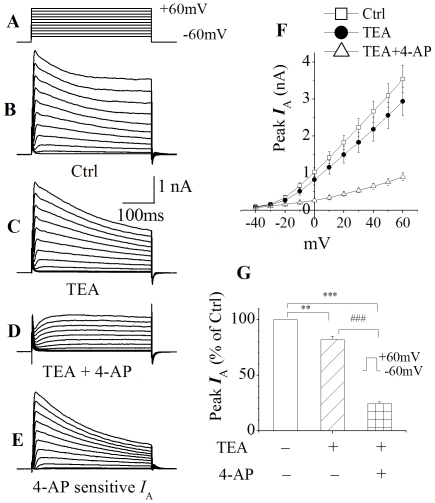
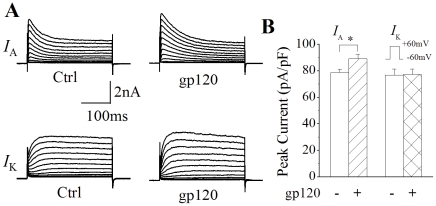
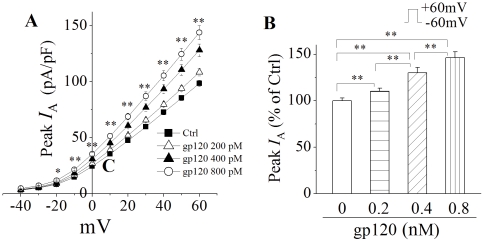
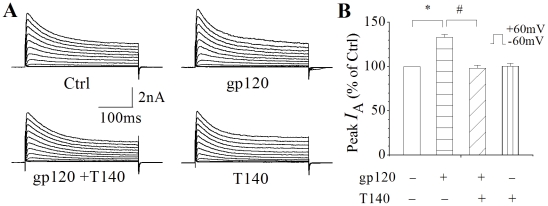
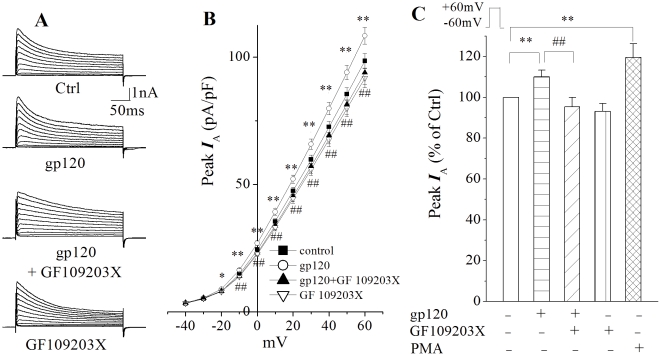
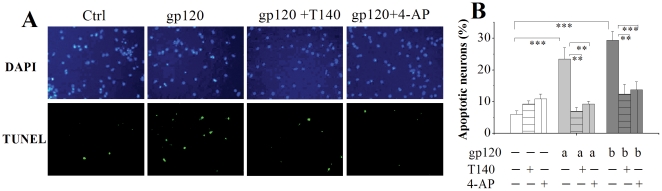
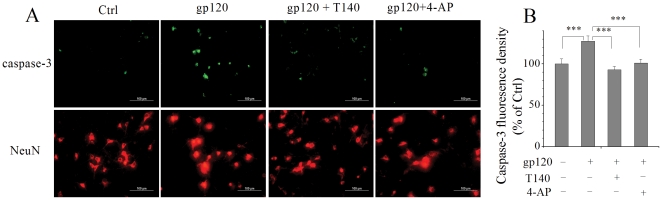
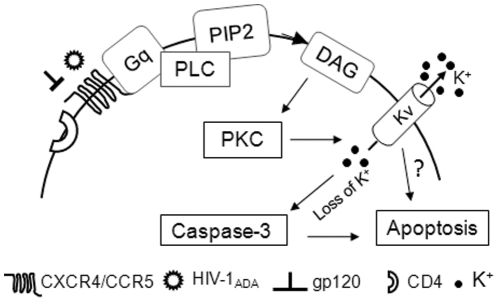
Human immunodeficiency virus type 1 (HIV-1)-associated dementia (HAD) usually occurs late in the course of HIV-1 infection and the mechanisms underlying HAD pathogenesis are not well understood. Accumulating evidence indicates that neuronal voltage-gated potassium (Kv) channels play an important role in memory processes and acquired neuronal channelopathies in HAD. To examine whether Kv channels are involved in HIV-1-associated neuronal injury, we studied the effects of HIV-1 glycoprotein 120 (gp120) on outward K+ currents in rat cortical neuronal cultures using whole-cell patch techniques. Exposure of cortical neurons to gp120 produced a dose-dependent enhancement of A-type transient outward K+ currents (IA). The gp120-induced increase of IA was attenuated by T140, a specific antagonist for chemokine receptor CXCR4, suggesting gp120 enhancement of neuronal IA via CXCR4. Pretreatment of neuronal cultures with a protein kinase C (PKC) inhibitor, GF109203X, inhibited the gp120-induced increase of IA. Biological significance of gp120 enhancement of IA was demonstrated by experimental results showing that gp120-induced neuronal apoptosis, as detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay and caspase-3 staining, was attenuated by either an IA blocker 4-aminopyridine or a specific CXCR4 antagonist T140. Taken together, these results suggest that gp120 may induce caspase-3 dependent neuronal apoptosis by enhancing IA via CXCR4-PKC signaling.
Conflict of interest statement
Figures
References
-
- McArthur JC, Brew BJ, Nath A. Neurological complications of HIV infection. Lancet Neurol. 2005;4:543–555. - PubMed
-
- Masliah E, Heaton RK, Marcotte TD, Ellis RJ, Wiley CA, et al. Dendritic injury is a pathological substrate for human immunodeficiency virus-related cognitive disorders. HNRC Group. The HIV Neurobehavioral Research Center. Ann Neurol. 1997;42:963–972. - PubMed
-
- Masliah E, Ge N, Achim C, Hansen L, Wiley C. Selective neuronal vulnerability in HIV encephalitis. J Neuropathol Exp Neurol. 1992;51:585–593. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
