

Biochemical analyses are instrumental in identifying the impact of mutations on holo and/or apo-forms and on the region(s) of alanine:glyoxylate aminotransferase variants associated with primary hyperoxaluria type I
- PMID: 22018727
- PMCID: PMC3271384
- DOI: 10.1016/j.ymgme.2011.09.033
Biochemical analyses are instrumental in identifying the impact of mutations on holo and/or apo-forms and on the region(s) of alanine:glyoxylate aminotransferase variants associated with primary hyperoxaluria type I
Abstract
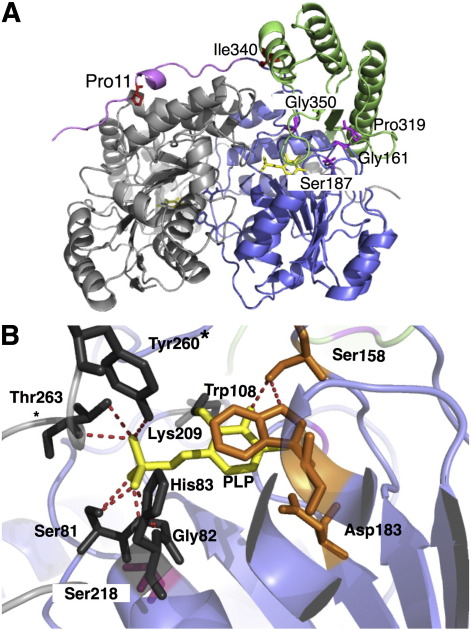
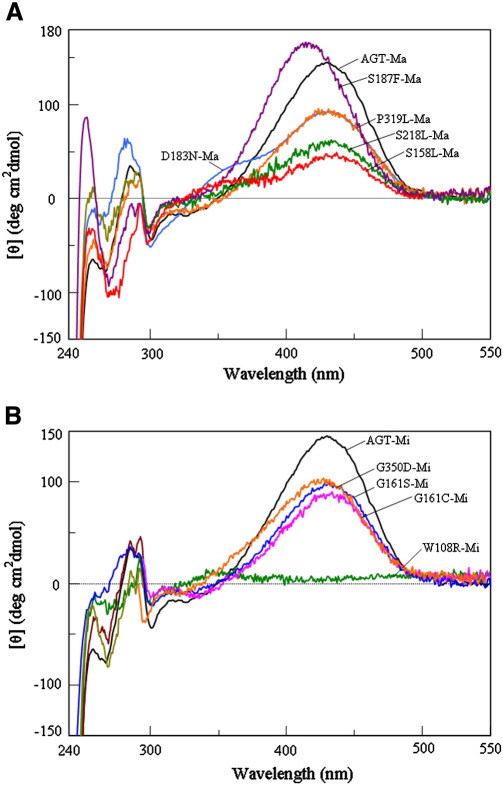
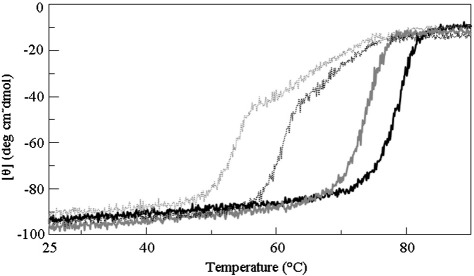
Primary Hyperoxaluria Type I (PH1) is a disorder of glyoxylate metabolism caused by mutations in the human AGXT gene encoding liver peroxisomal alanine:glyoxylate aminotransferase (AGT), a pyridoxal 5'-phosphate (PLP) dependent enzyme. Previous investigations highlighted that, although PH1 is characterized by a significant variability in terms of enzymatic phenotype, the majority of the pathogenic variants are believed to share both structural and functional defects, as mainly revealed by data on AGT activity and expression level in crude cellular extracts. However, the knowledge of the defects of the AGT variants at a protein level is still poor. We therefore performed a side-by-side comparison between normal AGT and nine purified recombinant pathogenic variants in terms of catalytic activity, coenzyme binding mode and affinity, spectroscopic features, oligomerization, and thermal stability of both the holo- and apo-forms. Notably, we chose four variants in which the mutated residues are located in the large domain of AGT either within the active site and interacting with the coenzyme or in its proximity, and five variants in which the mutated residues are distant from the active site either in the large or in the small domain. Overall, this integrated analysis of enzymatic activity, spectroscopic and stability information is used to (i) reassess previous data obtained with crude cellular extracts, (ii) establish which form(s) (i.e. holoenzyme and/or apoenzyme) and region(s) (i.e. active site microenvironment, large and/or small domain) of the protein are affected by each mutation, and (iii) suggest the possible therapeutic approach for patients bearing the examined mutations.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
References
-
- Danpure C.J., Rumsby G. Molecular aetiology of primary hyperoxaluria and its implications for clinical management. Expert Rev. Mol. Med. 2004;6:1–16. - PubMed
-
- Danpure C.J., Jennings P.R. Peroxisomal alanine:glyoxylate aminotransferase deficiency in primary hyperoxaluria type I. FEBS Lett. 1986;201:20–24. - PubMed
-
- Purdue P.E., Lumb M.J., Fox M., Griffo G., Hamon-Benais C., Povey S., Danpure C.J. Characterization and chromosomal mapping of a genomic clone encoding human alanine:glyoxylate aminotransferase. Genomics. 1991;10:34–42. - PubMed
-
- Purdue P.E., Lumb M.J., Allsop J., Danpure C.J. An intronic duplication in the alanine: glyoxylate aminotransferase gene facilitates identification of mutations in compound heterozygote patients with primary hyperoxaluria type 1. Hum. Genet. 1991;87:394–396. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
