

Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme
- PMID: 22020333
- PMCID: PMC3774279
- DOI: 10.1038/onc.2011.474
Acquired MET expression confers resistance to EGFR inhibition in a mouse model of glioblastoma multiforme
Abstract
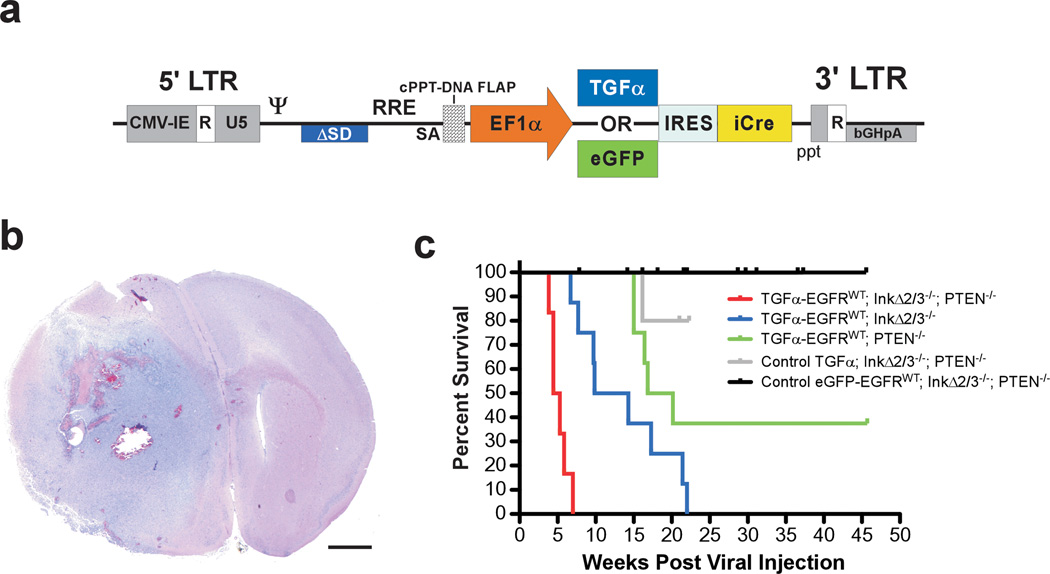
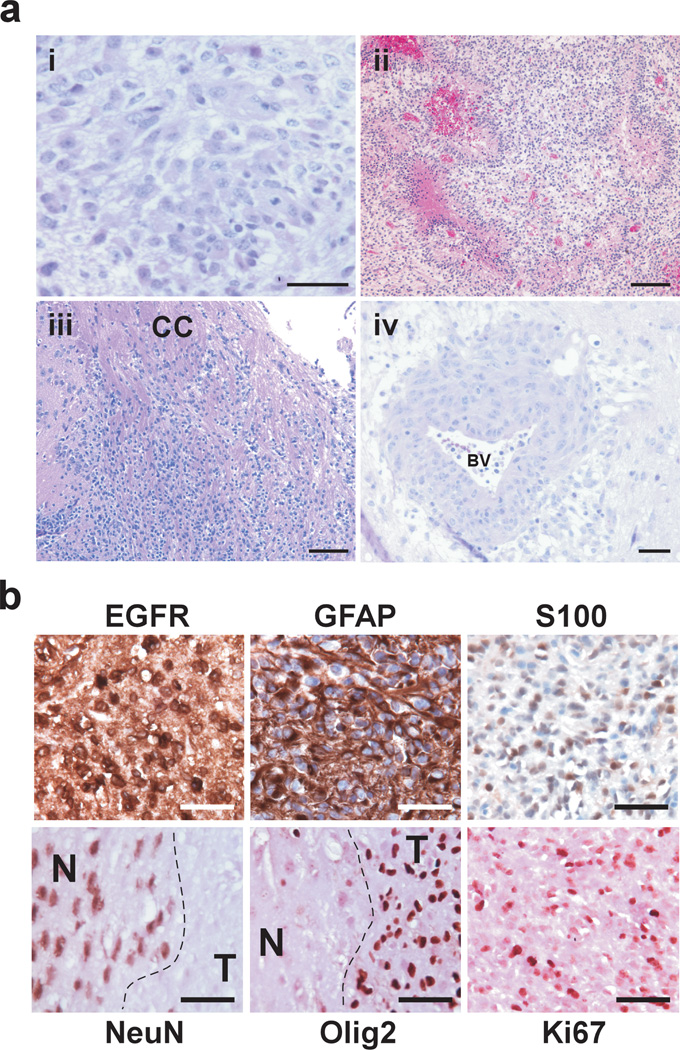
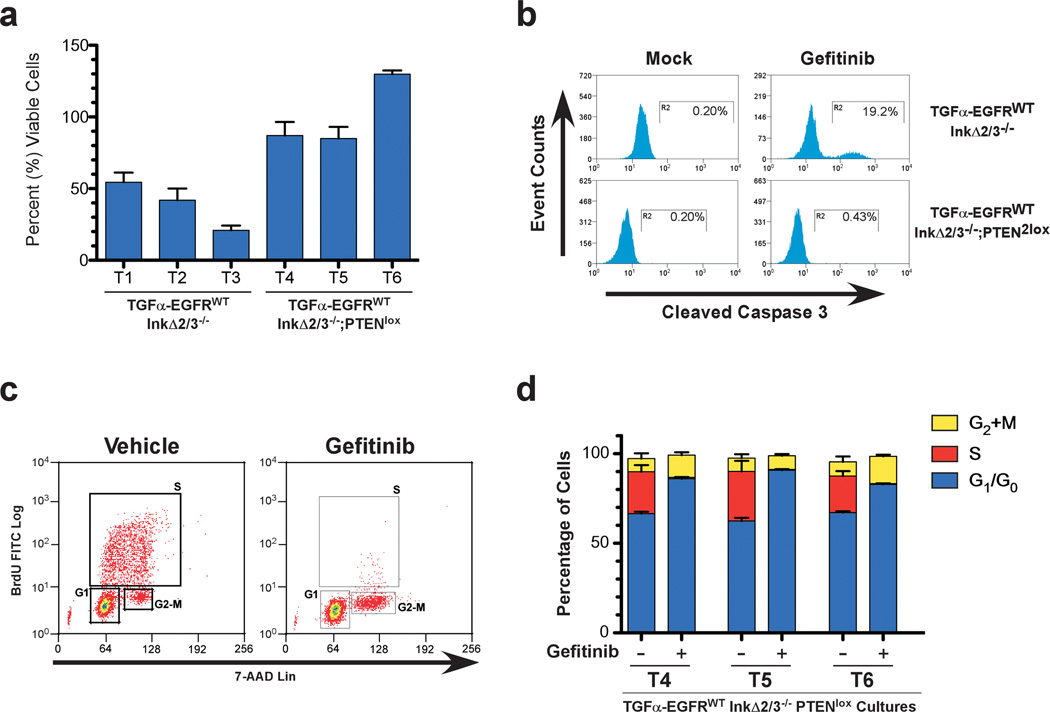
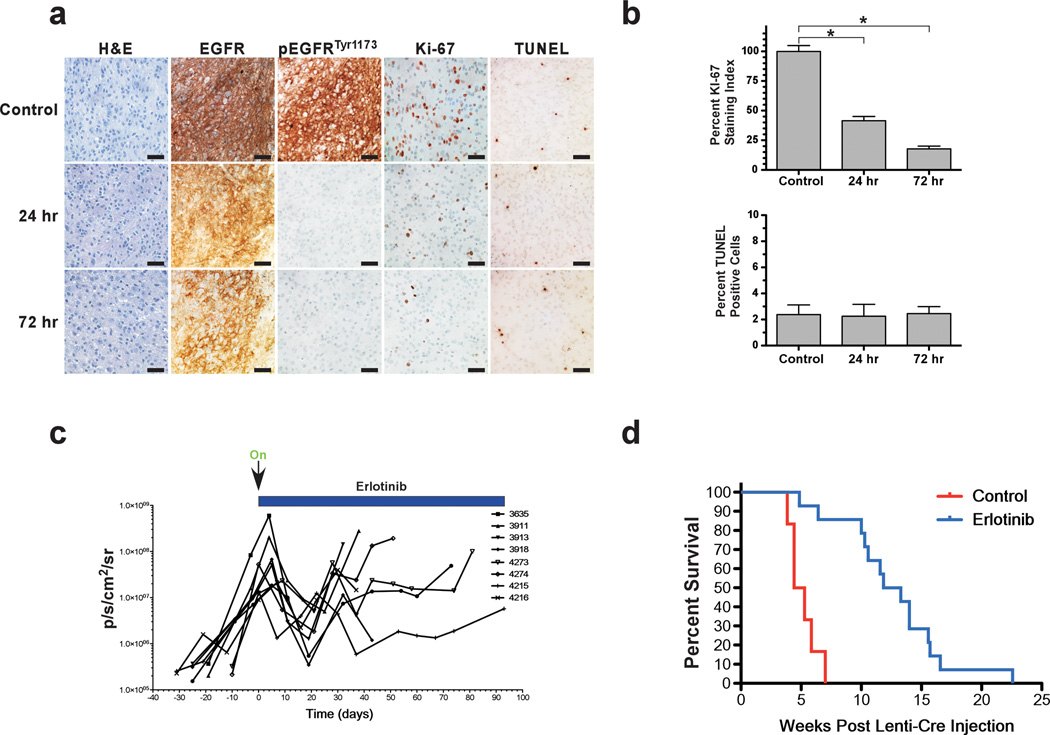
Glioblastoma multiforme (GBM) is an aggressive brain tumor for which there is no cure. Overexpression of wild-type epidermal growth factor receptor (EGFR) and loss of the tumor suppressor genes Ink4a/Arf and PTEN are salient features of this deadly cancer. Surprisingly, targeted inhibition of EGFR has been clinically disappointing, demonstrating an innate ability for GBM to develop resistance. Efforts at modeling GBM in mice using wild-type EGFR have proven unsuccessful to date, hampering endeavors at understanding molecular mechanisms of therapeutic resistance. Here, we describe a unique genetically engineered mouse model of EGFR-driven gliomagenesis that uses a somatic conditional overexpression and chronic activation of wild-type EGFR in cooperation with deletions in the Ink4a/Arf and PTEN genes in adult brains. Using this model, we establish that chronic activation of wild-type EGFR with a ligand is necessary for generating tumors with histopathological and molecular characteristics of GBMs. We show that these GBMs are resistant to EGFR kinase inhibition and we define this resistance molecularly. Inhibition of EGFR kinase activity using tyrosine kinase inhibitors in GBM tumor cells generates a cytostatic response characterized by a cell cycle arrest, which is accompanied by a substantial change in global gene expression levels. We demonstrate that an important component of this pattern is the transcriptional activation of the MET receptor tyrosine kinase and that pharmacological inhibition of MET overcomes the resistance to EGFR inhibition in these cells. These findings provide important new insights into mechanisms of resistance to EGFR inhibition and suggest that inhibition of multiple targets will be necessary to provide therapeutic benefit for GBM patients.
Conflict of interest statement
The authors have declared that no conflict of interest exists.
Figures
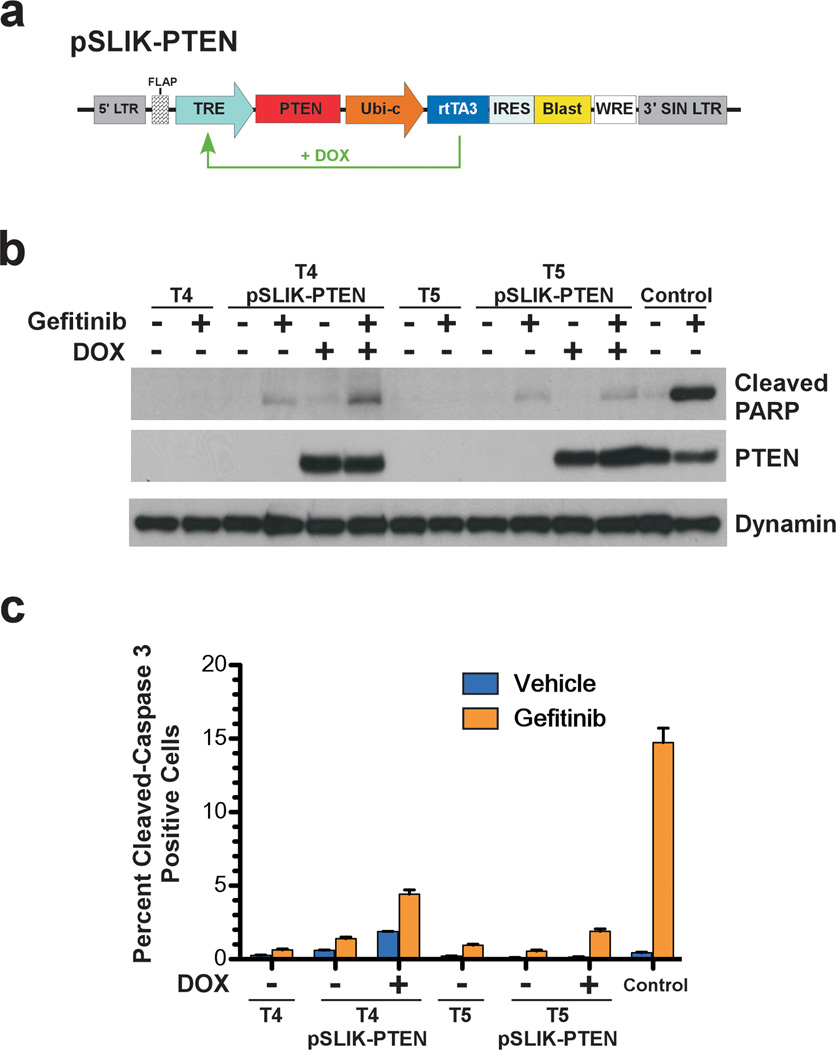
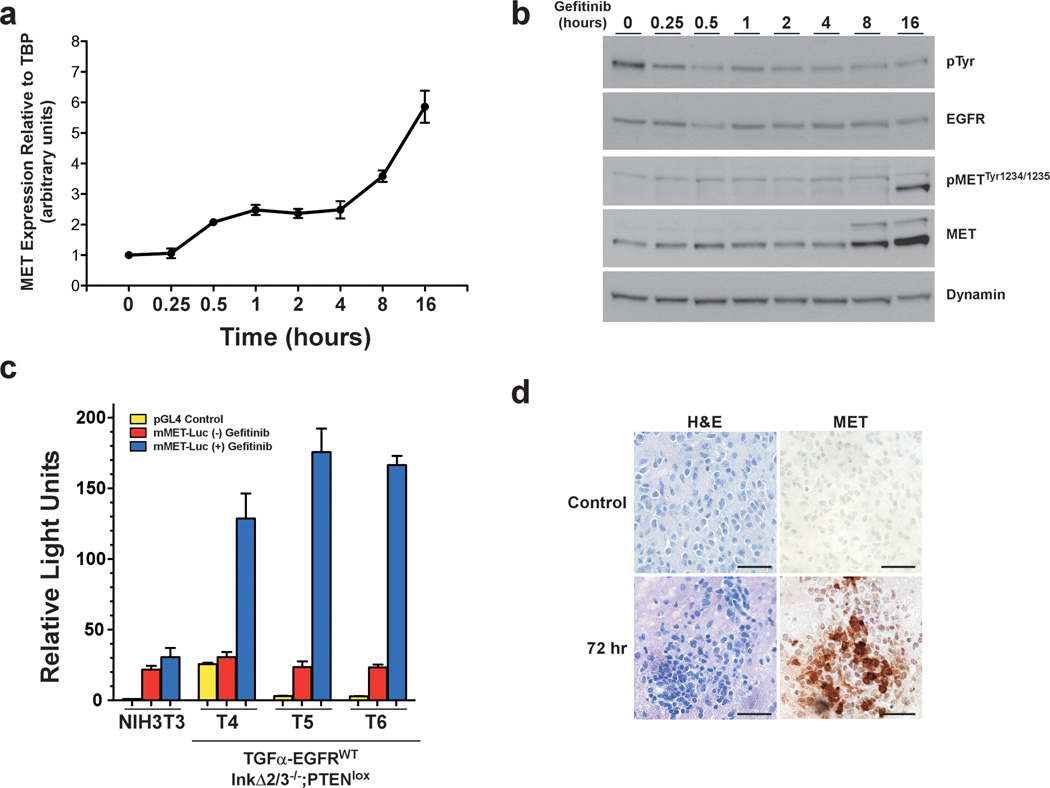
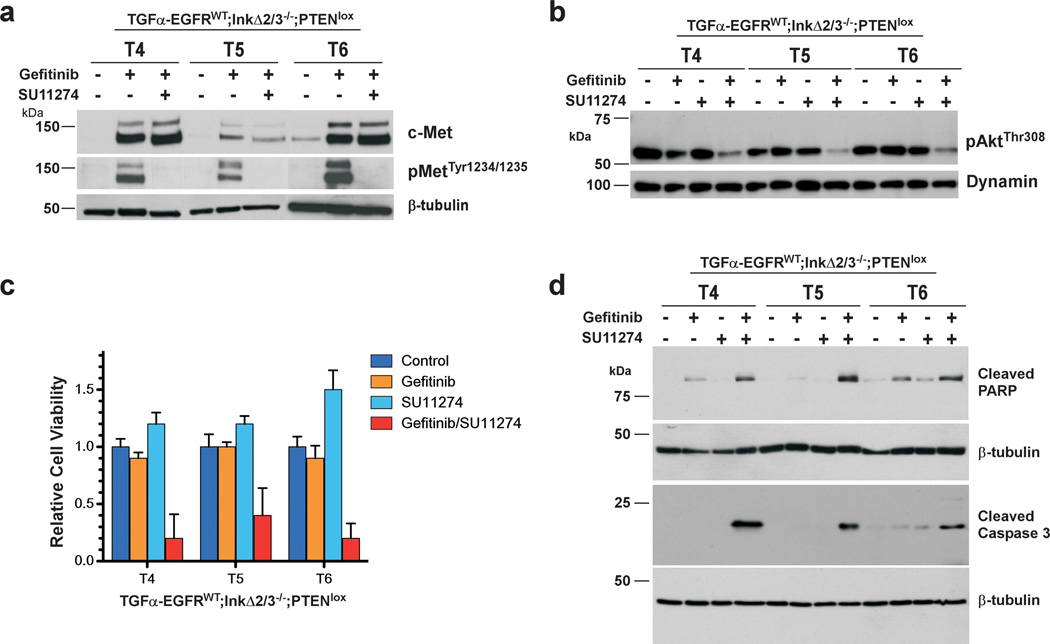
References
-
- Andl CD, Mizushima T, Nakagawa H, Oyama K, Harada H, Chruma K, et al. Epidermal growth factor receptor mediates increased cell proliferation, migration, and aggregation in esophageal keratinocytes in vitro and in vivo. J Biol Chem. 2003;278:1824–1830. - PubMed
-
- Busse D, Doughty RS, Ramsey TT, Russell WE, Price JO, Flanagan WM, et al. Reversible G(1) arrest induced by inhibition of the epidermal growth factor receptor tyrosine kinase requires up-regulation of p27(KIP1) independent of MAPK activity. J Biol Chem. 2000;275:6987–6995. - PubMed
-
- Cheng J, Huang H, Zhang ZT, Shapiro E, Pellicer A, Sun TT, et al. Overexpression of epidermal growth factor receptor in urothelium elicits urothelial hyperplasia and promotes bladder tumor growth. Cancer Res. 2002;62:4157–4163. - PubMed
-
- Ciardiello F, McGeady ML, Kim N, Basolo F, Hynes N, Langton BC, et al. Transforming growth factor-alpha expression is enhanced in human mammary epithelial cells transformed by an activated c-Ha-ras protooncogene but not by the c-neu protooncogene, and overexpression of the transforming growth factor-alpha complementary DNA leads to transformation. Cell Growth Differ. 1990;1:407–420. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
