

Regulation of Cdk7 activity through a phosphatidylinositol (3)-kinase/PKC-ι-mediated signaling cascade in glioblastoma
- PMID: 22021906
- PMCID: PMC4662058
- DOI: 10.1093/carcin/bgr231
Regulation of Cdk7 activity through a phosphatidylinositol (3)-kinase/PKC-ι-mediated signaling cascade in glioblastoma
Abstract
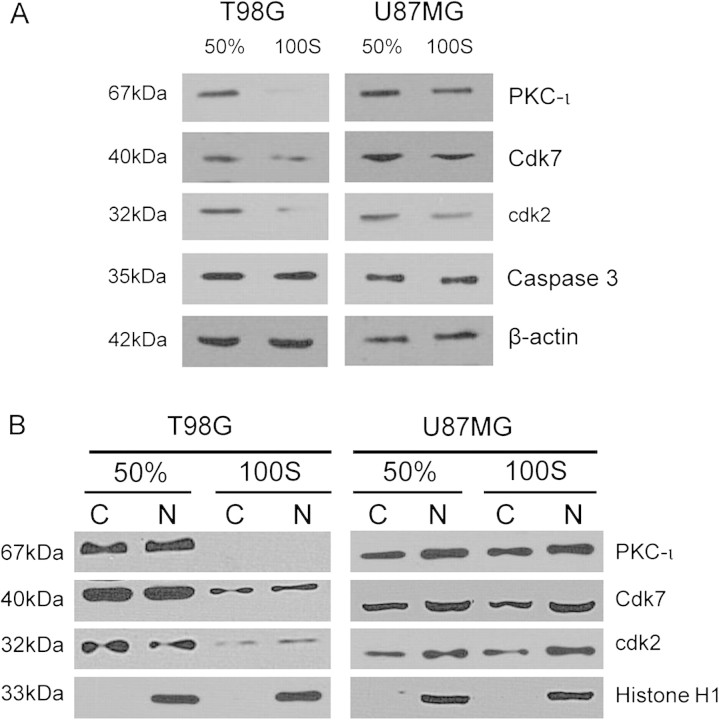
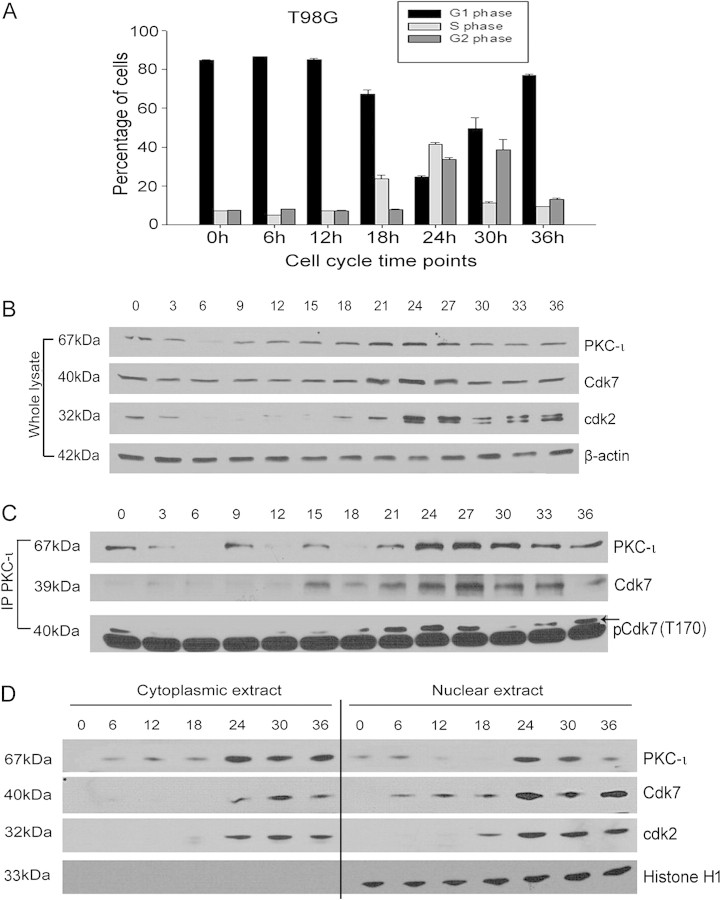
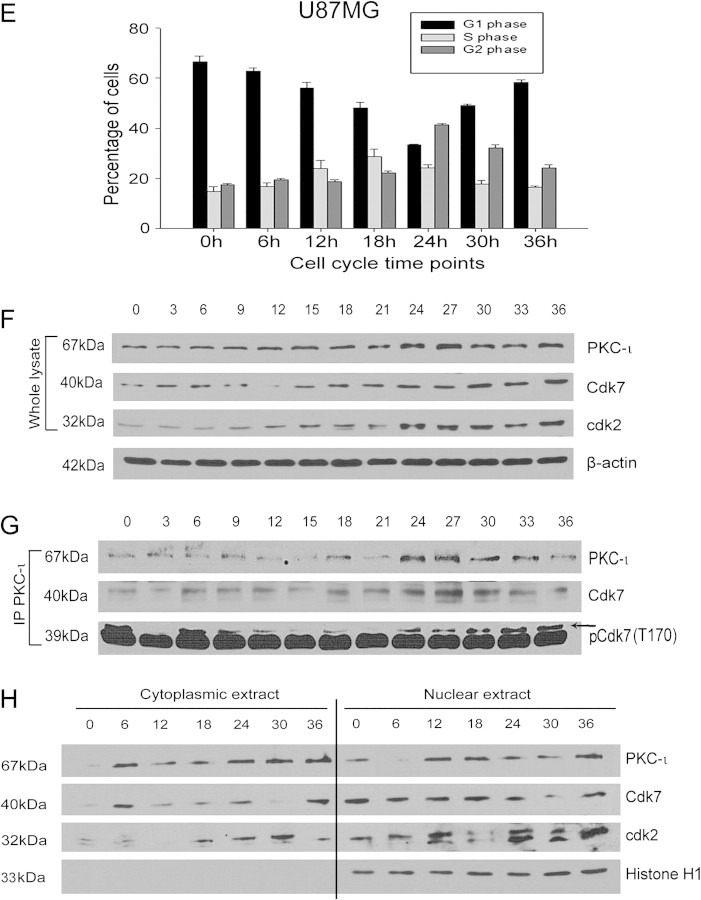
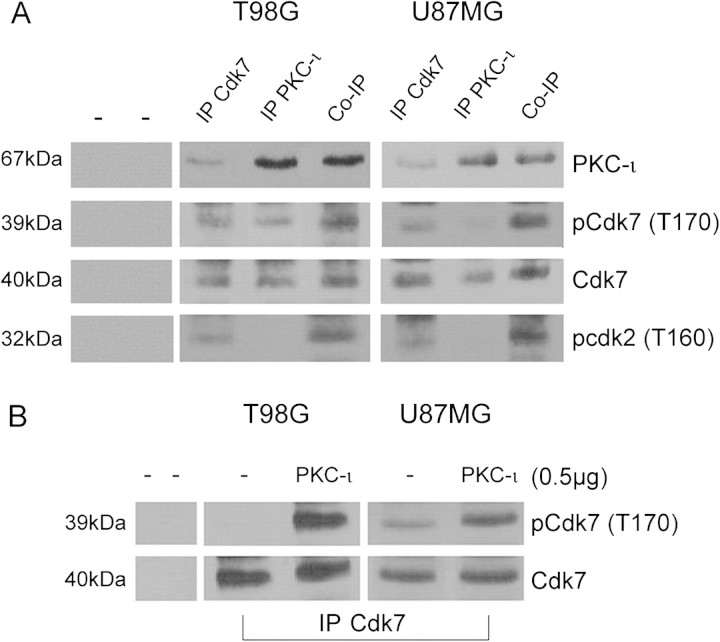
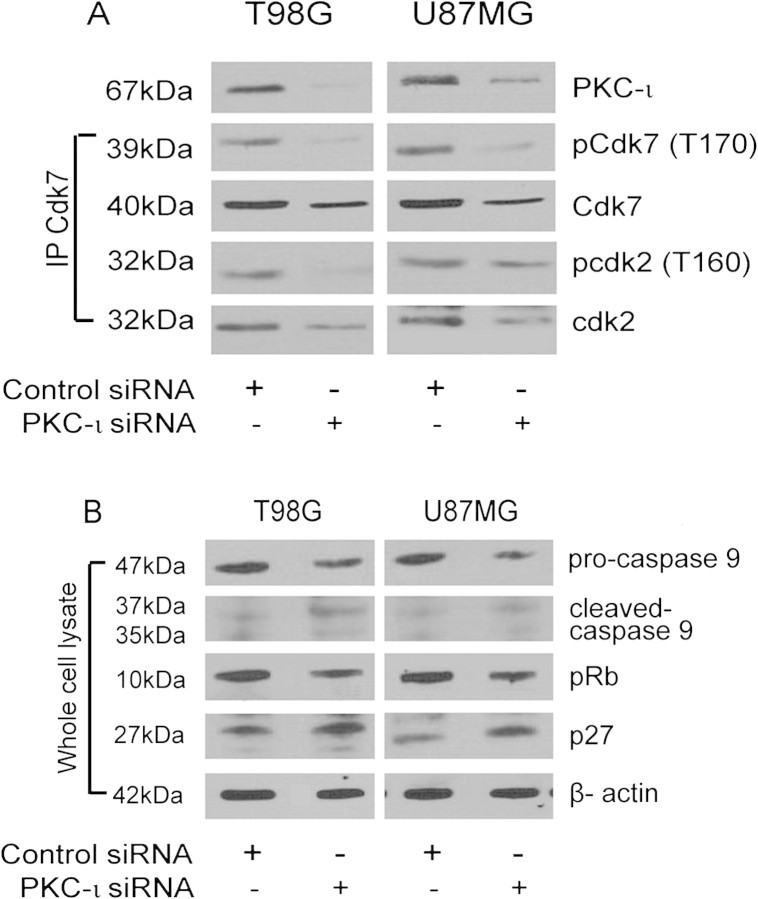
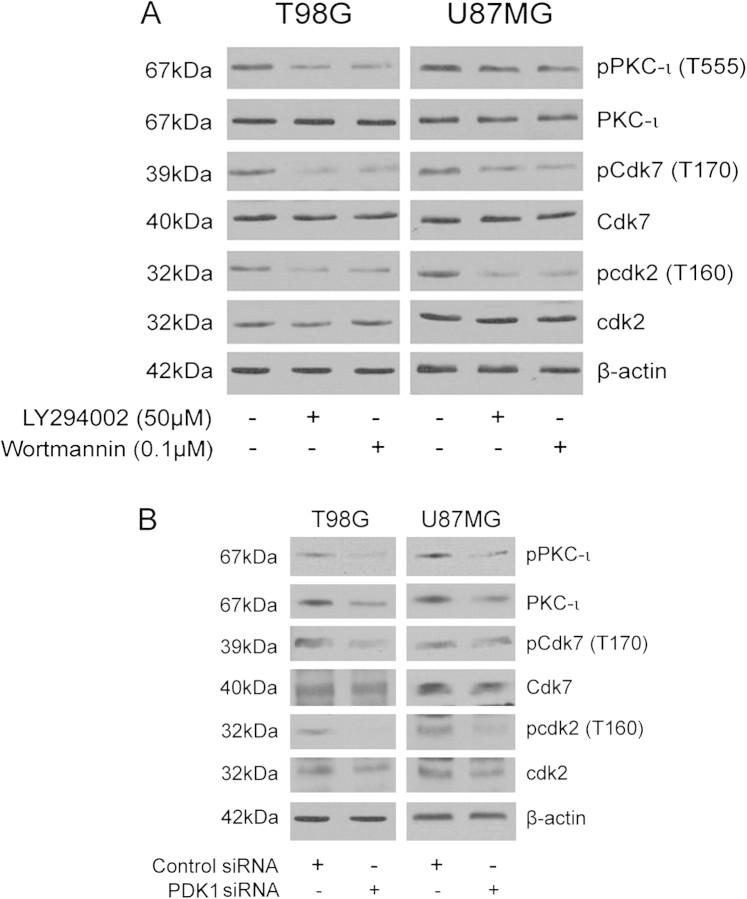
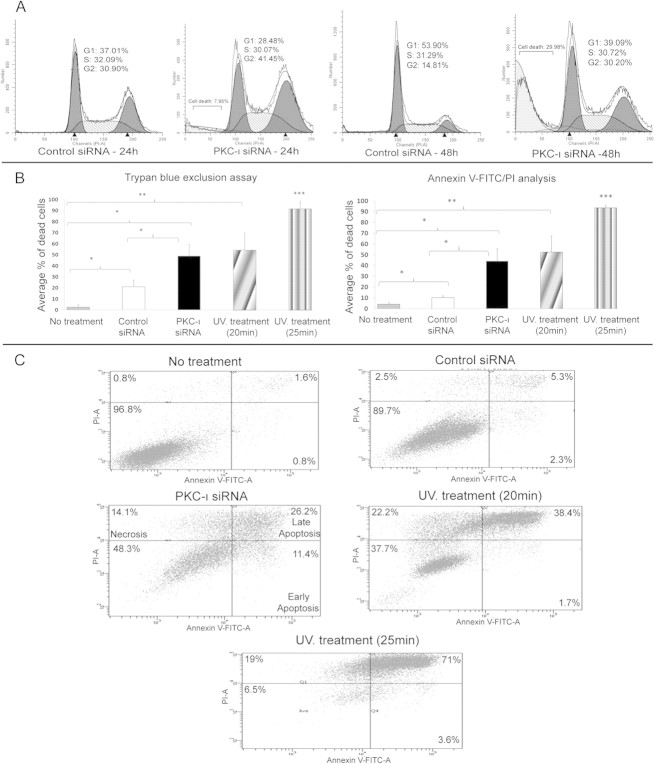
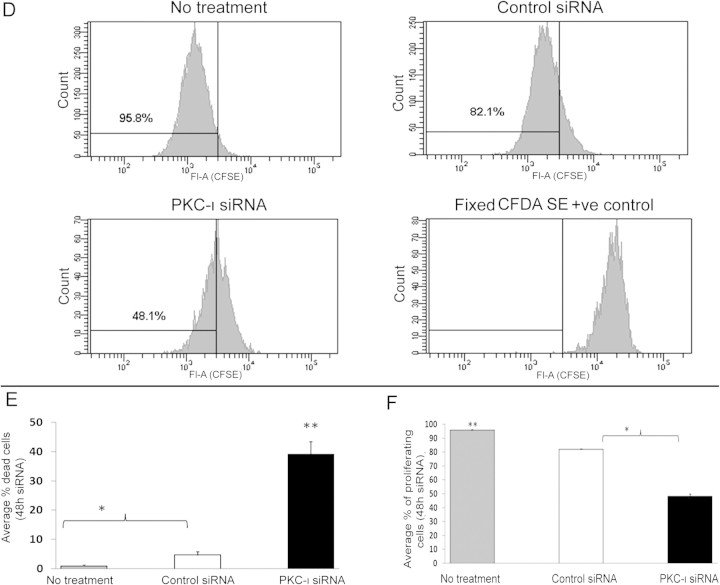
The objective of this research was to study the potential function of protein kinase C (PKC)-ι in cell cycle progression and proliferation in glioblastoma. PKC-ι is highly overexpressed in human glioma and benign and malignant meningioma; however, little is understood about its role in regulating cell proliferation of glioblastoma. Several upstream molecular aberrations and/or loss of PTEN have been implicated to constitutively activate the phosphatidylinositol (PI) (3)-kinase pathway. PKC-ι is a targeted mediator in the PI (3)-kinase signal transduction repertoire. Results showed that PKC-ι was highly activated and overexpressed in glioma cells. PKC-ι directly associated and phosphorylated Cdk7 at T170 in a cell cycle-dependent manner, phosphorylating its downstream target, cdk2 at T160. Cdk2 has a major role in inducing G(1)-S phase progression of cells. Purified PKC-ι phosphorylated both endogenous and exogenous Cdk7. PKC-ι downregulation reduced Cdk7 and cdk2 phosphorylation following PI (3)-kinase inhibition, phosphotidylinositol-dependent kinase 1 knockdown as well as PKC-ι silencing (by siRNA treatment). It also diminished cdk2 activity. PKC-ι knockdown inhibited overall proliferation rates and induced apoptosis in glioma cells. These findings suggest that glioma cells may be proliferating through a novel PI (3)-kinase-/PKC-ι/Cdk7/cdk2-mediated pathway.
Figures
Similar articles
-
Human glioma PKC-iota and PKC-betaII phosphorylate cyclin-dependent kinase activating kinase during the cell cycle.Cell Prolif. 2002 Feb;35(1):23-36. doi: 10.1046/j.1365-2184.2002.00220.x. Cell Prolif. 2002. PMID: 11856176 Free PMC article.
-
Role of protein kinase C-iota in transformed non-malignant RWPE-1 cells and androgen-independent prostate carcinoma DU-145 cells.Cell Prolif. 2009 Apr;42(2):182-94. doi: 10.1111/j.1365-2184.2009.00582.x. Cell Prolif. 2009. PMID: 19243387 Free PMC article.
-
PKC-ι promotes glioblastoma cell survival by phosphorylating and inhibiting BAD through a phosphatidylinositol 3-kinase pathway.Biochim Biophys Acta. 2011 Jun;1813(6):1190-7. doi: 10.1016/j.bbamcr.2011.03.007. Epub 2011 Mar 17. Biochim Biophys Acta. 2011. PMID: 21419810
-
Linking protein kinase C to cell-cycle control.Eur J Biochem. 1997 Aug 15;248(1):1-9. doi: 10.1111/j.1432-1033.1997.t01-4-00001.x. Eur J Biochem. 1997. PMID: 9310352 Review.
-
PKC signaling in glioblastoma.Cancer Biol Ther. 2013 Apr;14(4):287-94. doi: 10.4161/cbt.23615. Epub 2013 Jan 28. Cancer Biol Ther. 2013. PMID: 23358475 Free PMC article. Review.
Cited by
-
A patient tumor transplant model of squamous cell cancer identifies PI3K inhibitors as candidate therapeutics in defined molecular bins.Mol Oncol. 2013 Aug;7(4):776-90. doi: 10.1016/j.molonc.2013.03.004. Epub 2013 Apr 4. Mol Oncol. 2013. PMID: 23607916 Free PMC article.
-
Serum cyclin-dependent kinase 9 is a potential biomarker of atherosclerotic inflammation.Oncotarget. 2016 Jan 12;7(2):1854-62. doi: 10.18632/oncotarget.6443. Oncotarget. 2016. PMID: 26636538 Free PMC article.
-
A high-throughput screen identifies that CDK7 activates glucose consumption in lung cancer cells.Nat Commun. 2019 Nov 29;10(1):5444. doi: 10.1038/s41467-019-13334-8. Nat Commun. 2019. PMID: 31784510 Free PMC article.
-
Protein Kinase C (PKC) Isozymes as Diagnostic and Prognostic Biomarkers and Therapeutic Targets for Cancer.Cancers (Basel). 2022 Nov 3;14(21):5425. doi: 10.3390/cancers14215425. Cancers (Basel). 2022. PMID: 36358843 Free PMC article. Review.
-
CDK7 in breast cancer: mechanisms of action and therapeutic potential.Cell Commun Signal. 2024 Apr 11;22(1):226. doi: 10.1186/s12964-024-01577-y. Cell Commun Signal. 2024. PMID: 38605321 Free PMC article. Review.
References
-
- Parlato C, et al. Surgery, radiotherapy and temozolomide in treating high-grade gliomas. Front. Biosci. 2006;11:1280–1283. - PubMed
-
- Stupp R, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005;352:987–996. - PubMed
-
- Xiao H, et al. The identification of four protein kinase C isoforms in human glioblastoma cell line: PKC alpha, gamma, epsilon and zeta. J. Neurosurg. 1994;81:734–740. - PubMed
-
- Couldwell W, et al. Inhibition of growth of established human glioma cell lines by modulators of the protein kinase C system. J. Neurosurg. 1990;73:594–600. - PubMed
-
- Inoue M, et al. Studies on cyclic nucleotide-independent protein kinase and its proenzyme in mammalian tissues. II. Proenzyme and its activation by calcium—dependent protease from rat brain. J. Biol. Chem. 1977;252:7610–7616. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
