

Distinct epigenomic features in end-stage failing human hearts
- PMID: 22025602
- PMCID: PMC3634158
- DOI: 10.1161/CIRCULATIONAHA.111.040071
Distinct epigenomic features in end-stage failing human hearts
Abstract
Background: The epigenome refers to marks on the genome, including DNA methylation and histone modifications, that regulate the expression of underlying genes. A consistent profile of gene expression changes in end-stage cardiomyopathy led us to hypothesize that distinct global patterns of the epigenome may also exist.
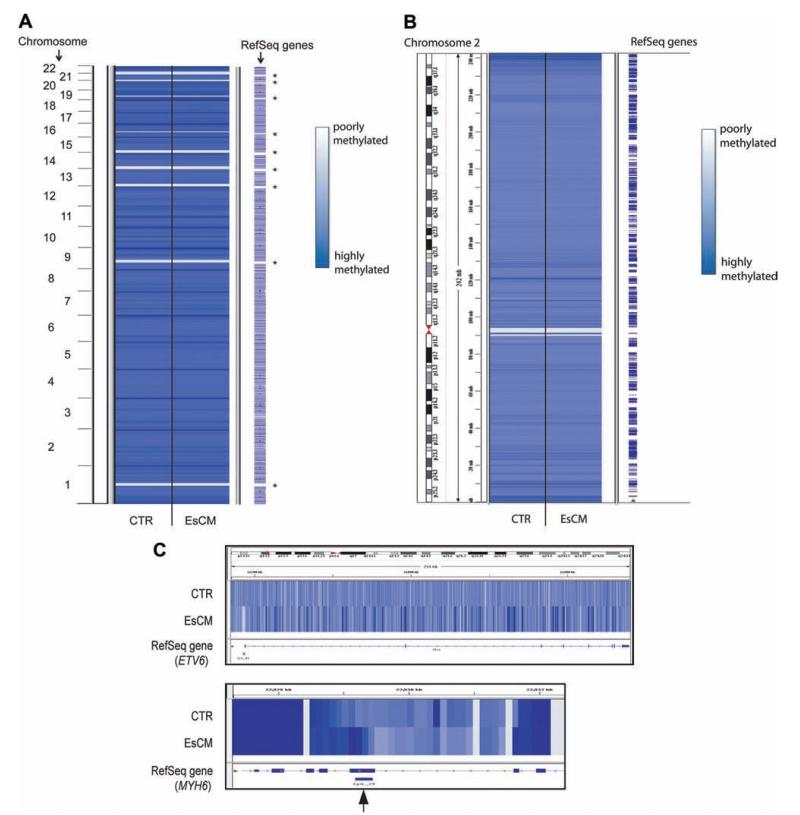
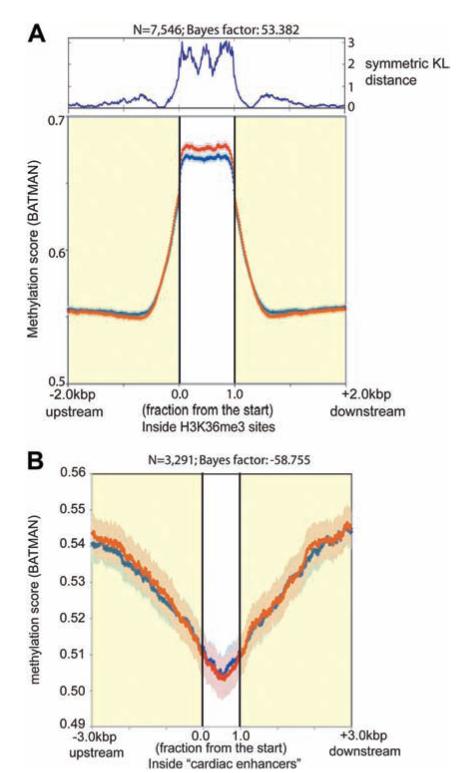
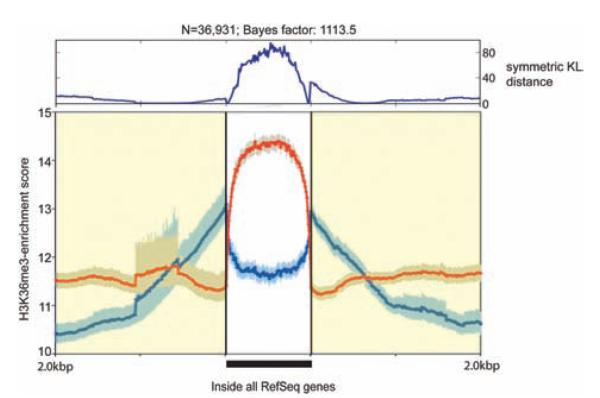
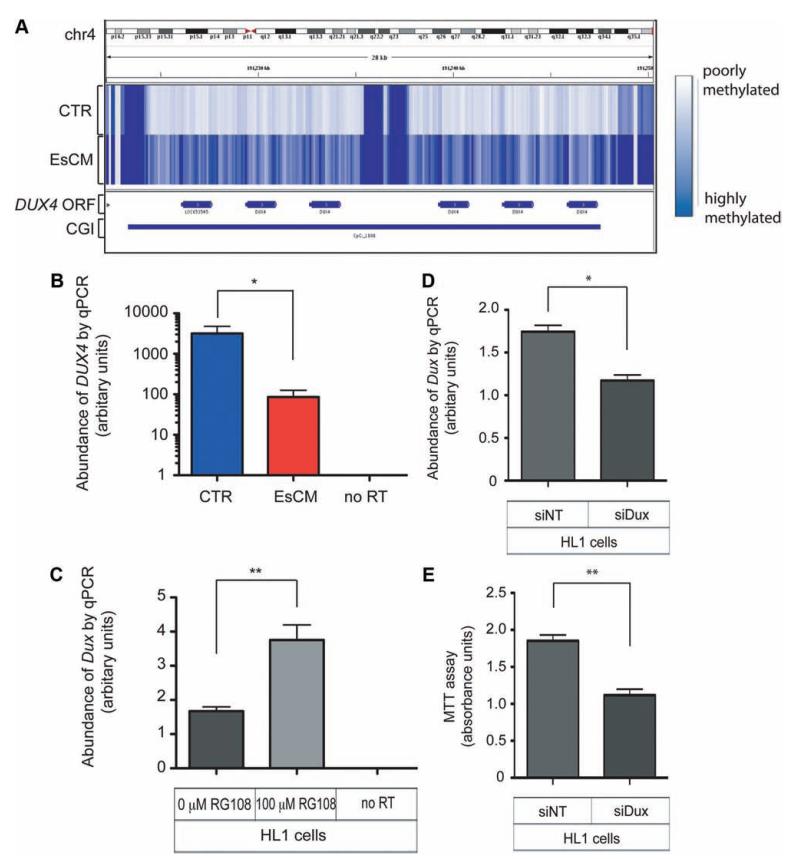
Methods and results: We constructed genome-wide maps of DNA methylation and histone-3 lysine-36 trimethylation (H3K36me3) enrichment for cardiomyopathic and normal human hearts. More than 506 Mb sequences per library were generated by high-throughput sequencing, allowing us to assign methylation scores to ≈28 million CG dinucleotides in the human genome. DNA methylation was significantly different in promoter CpG islands, intragenic CpG islands, gene bodies, and H3K36me3-enriched regions of the genome. DNA methylation differences were present in promoters of upregulated genes but not downregulated genes. H3K36me3 enrichment itself was also significantly different in coding regions of the genome. Specifically, abundance of RNA transcripts encoded by the DUX4 locus correlated to differential DNA methylation and H3K36me3 enrichment. In vitro, Dux gene expression was responsive to a specific inhibitor of DNA methyltransferase, and Dux siRNA knockdown led to reduced cell viability.
Conclusions: Distinct epigenomic patterns exist in important DNA elements of the cardiac genome in human end-stage cardiomyopathy. The epigenome may control the expression of local or distal genes with critical functions in myocardial stress response. If epigenomic patterns track with disease progression, assays for the epigenome may be useful for assessing prognosis in heart failure. Further studies are needed to determine whether and how the epigenome contributes to the development of cardiomyopathy.
Figures


References
-
- Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. - PubMed
-
- Dorn GW, 2nd, Matkovich SJ. Put your chips on transcriptomics. Circulation. 2008;118:216–218. - PubMed
-
- Creemers EE, Wilde AA, Pinto YM. Heart failure: advances through genomics. Nat Rev Genet. 2011;12:357–362. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
