

A cardiac pathway of cyclic GMP-independent signaling of guanylyl cyclase A, the receptor for atrial natriuretic peptide
- PMID: 22027011
- PMCID: PMC3215055
- DOI: 10.1073/pnas.1103300108
A cardiac pathway of cyclic GMP-independent signaling of guanylyl cyclase A, the receptor for atrial natriuretic peptide
Abstract
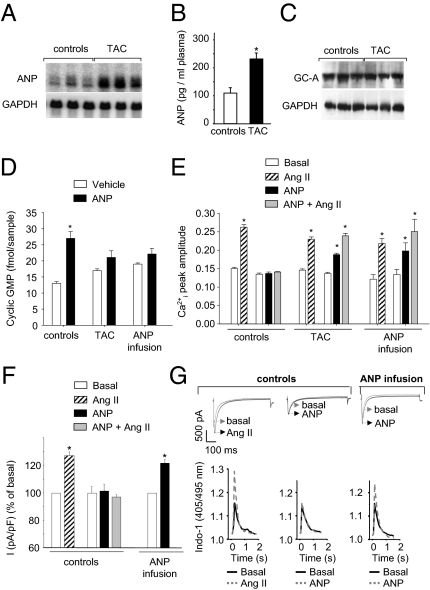
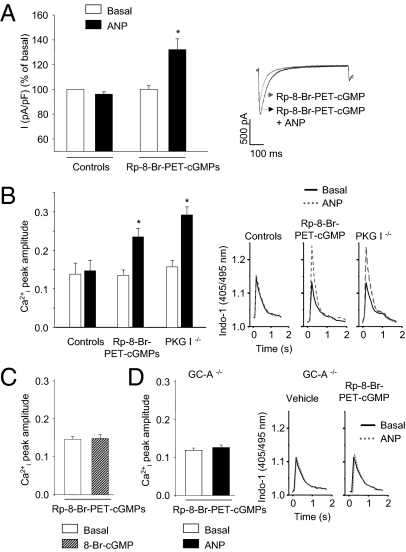
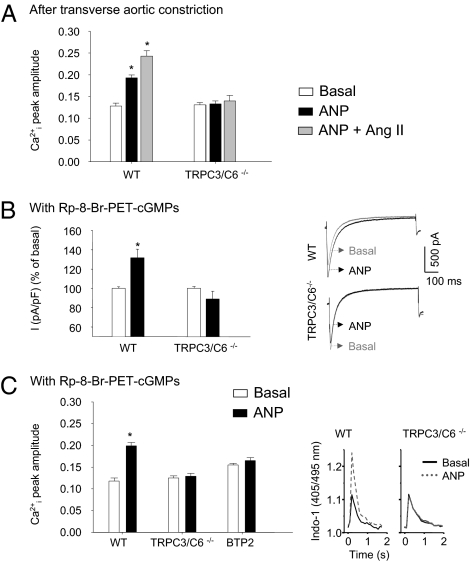
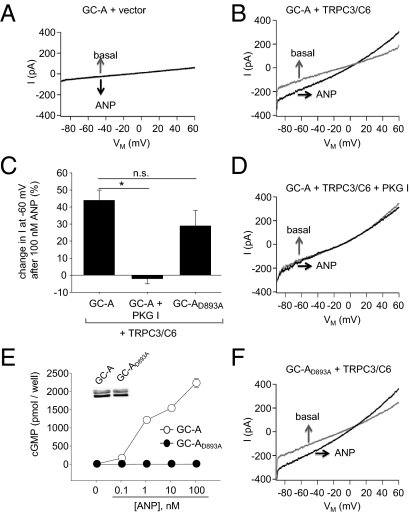
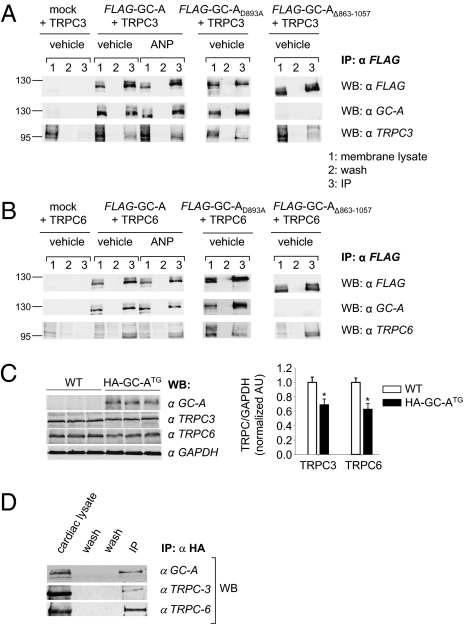
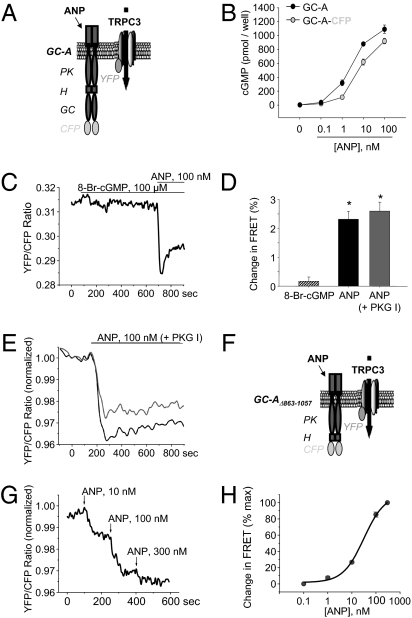
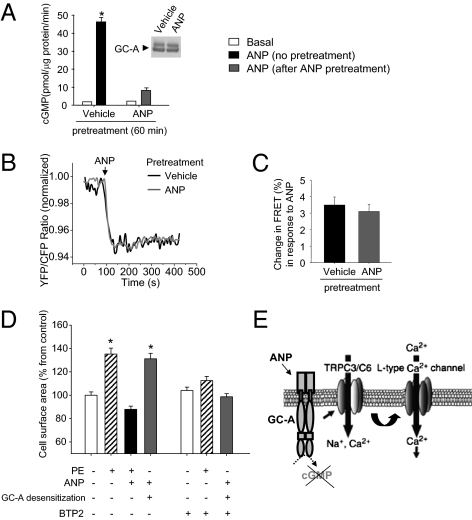
Cardiac atrial natriuretic peptide (ANP) regulates arterial blood pressure, moderates cardiomyocyte growth, and stimulates angiogenesis and metabolism. ANP binds to the transmembrane guanylyl cyclase (GC) receptor, GC-A, to exert its diverse functions. This process involves a cGMP-dependent signaling pathway preventing pathological [Ca(2+)](i) increases in myocytes. In chronic cardiac hypertrophy, however, ANP levels are markedly increased and GC-A/cGMP responses to ANP are blunted due to receptor desensitization. Here we show that, in this situation, ANP binding to GC-A stimulates a unique cGMP-independent signaling pathway in cardiac myocytes, resulting in pathologically elevated intracellular Ca(2+) levels. This pathway involves the activation of Ca(2+)-permeable transient receptor potential canonical 3/6 (TRPC3/C6) cation channels by GC-A, which forms a stable complex with TRPC3/C6 channels. Our results indicate that the resulting cation influx activates voltage-dependent L-type Ca(2+) channels and ultimately increases myocyte Ca(2)(+)(i) levels. These observations reveal a dual role of the ANP/GC-A-signaling pathway in the regulation of cardiac myocyte Ca(2+)(i) homeostasis. Under physiological conditions, activation of a cGMP-dependent pathway moderates the Ca(2+)(i)-enhancing action of hypertrophic factors such as angiotensin II. By contrast, a cGMP-independent pathway predominates under pathophysiological conditions when GC-A is desensitized by high ANP levels. The concomitant rise in [Ca(2+)](i) might increase the propensity to cardiac hypertrophy and arrhythmias.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Kuhn M. Structure, regulation, and function of mammalian membrane guanylyl cyclase receptors, with a focus on guanylyl cyclase-A. Circ Res. 2003;93:700–709. - PubMed
-
- Kinoshita H, et al. Inhibition of TRPC6 channel activity contributes to the antihypertrophic effects of natriuretic peptides-guanylyl cyclase-A signaling in the heart. Circ Res. 2010;106:1849–1860. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
