

Neuroacanthocytosis syndromes
- PMID: 22027213
- PMCID: PMC3212896
- DOI: 10.1186/1750-1172-6-68
Neuroacanthocytosis syndromes
Abstract
Neuroacanthocytosis (NA) syndromes are a group of genetically defined diseases characterized by the association of red blood cell acanthocytosis and progressive degeneration of the basal ganglia. NA syndromes are exceptionally rare with an estimated prevalence of less than 1 to 5 per 1'000'000 inhabitants for each disorder. The core NA syndromes include autosomal recessive chorea-acanthocytosis and X-linked McLeod syndrome which have a Huntington's disease-like phenotype consisting of a choreatic movement disorder, psychiatric manifestations and cognitive decline, and additional multi-system features including myopathy and axonal neuropathy. In addition, cardiomyopathy may occur in McLeod syndrome. Acanthocytes are also found in a proportion of patients with autosomal dominant Huntington's disease-like 2, autosomal recessive pantothenate kinase-associated neurodegeneration and several inherited disorders of lipoprotein metabolism, namely abetalipoproteinemia (Bassen-Kornzweig syndrome) and hypobetalipoproteinemia leading to vitamin E malabsorption. The latter disorders are characterized by a peripheral neuropathy and sensory ataxia due to dorsal column degeneration, but movement disorders and cognitive impairment are not present. NA syndromes are caused by disease-specific genetic mutations. The mechanism by which these mutations cause neurodegeneration is not known. The association of the acanthocytic membrane abnormality with selective degeneration of the basal ganglia, however, suggests a common pathogenetic pathway. Laboratory tests include blood smears to detect acanthocytosis and determination of serum creatine kinase. Cerebral magnetic resonance imaging may demonstrate striatal atrophy. Kell and Kx blood group antigens are reduced or absent in McLeod syndrome. Western blot for chorein demonstrates absence of this protein in red blood cells of chorea-acanthocytosis patients. Specific genetic testing is possible in all NA syndromes. Differential diagnoses include Huntington disease and other causes of progressive hyperkinetic movement disorders. There are no curative therapies for NA syndromes. Regular cardiologic studies and avoidance of transfusion complications are mandatory in McLeod syndrome. The hyperkinetic movement disorder may be treated as in Huntington disease. Other symptoms including psychiatric manifestations should be managed in a symptom-oriented manner. NA syndromes have a relentlessly progressive course usually over two to three decades.
Figures
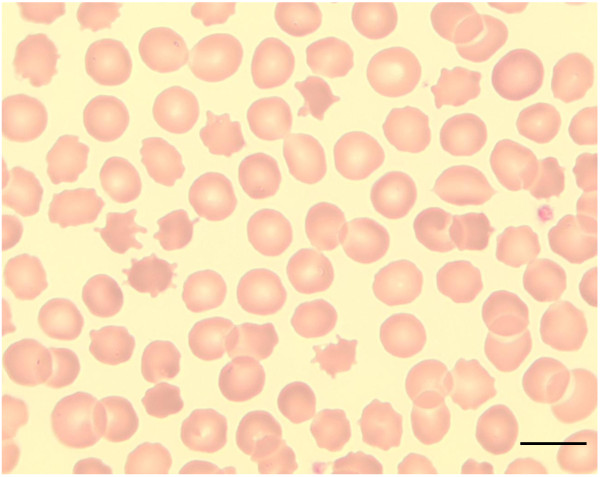
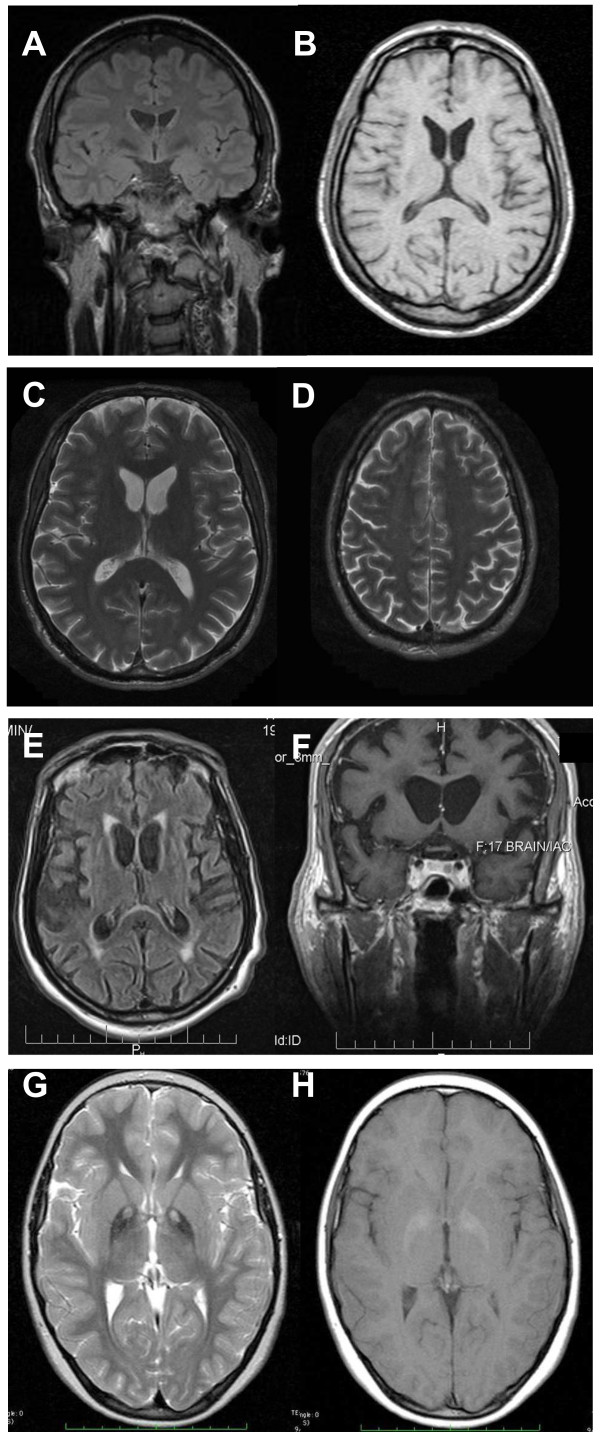
References
-
- Walker RH, Jung HH, Dobson-Stone C, Rampoldi L, Sano A, Tison F, Danek A. Neurologic phenotypes associated with acanthocytosis. Neurology. 2007;68(2):92–98. doi: 10.1212/01.wnl.0000250356.78092.cc. - DOI - PubMed
-
- Critchley EM, Clark DB, Wikler A. Acanthocytosis and neurological disorder without betalipoproteinemia. ArchNeurol. 1968;18(2):134–140. - PubMed
-
- Levine IM, Estes JW, Looney JM. Hereditary neurological disease with acanthocytosis. A new syndrome. ArchNeurol. 1968;19(4):403–409. - PubMed
-
- Marsh WL, Taswell HF, Oyen R, Nichols ME, Vergara MS, Pineda AA. Kx antigen of the Kell system and its relationship to chronic granulomatous disease. Evidence the Kx gene is X-linked. Transfusion. 1975;15:527.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
