

Elucidating the mechanism of regulation of transforming growth factor β Type II receptor expression in human lung cancer cell lines
- PMID: 22028617
- PMCID: PMC3201568
- DOI: 10.1593/neo.11576
Elucidating the mechanism of regulation of transforming growth factor β Type II receptor expression in human lung cancer cell lines
Abstract
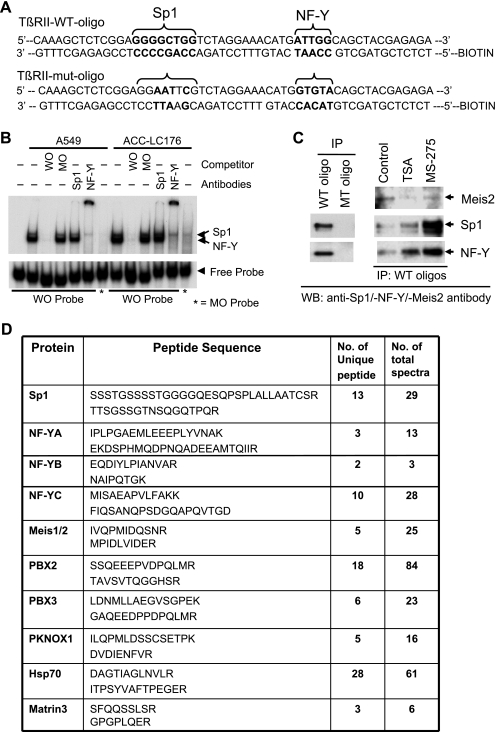
Lung carcinogenesis in humans involves an accumulation of genetic and epigenetic changes that lead to alterations in normal lung epithelium, to in situ carcinoma, and finally to invasive and metastatic cancers. The loss of transforming growth factor β (TGF-β)-induced tumor suppressor function in tumors plays a pivotal role in this process, and our previous studies have shown that resistance to TGF-β in lung cancers occurs mostly through the loss of TGF-β type II receptor expression (TβRII). However, little is known about the mechanism of down-regulation of TβRII and how histone deacetylase (HDAC) inhibitors (HDIs) can restore TGF-β-induced tumor suppressor function. Here we show that HDIs restore TβRII expression and that DNA hypermethylation has no effect on TβRII promoter activity in lung cancer cell lines. TGF-β-induced tumor suppressor function is restored by HDIs in lung cancer cell lines that lack TβRII expression. Activation of mitogen-activated protein kinase/extracellular signal-regulated kinase pathway by either activated Ras or epidermal growth factor signaling is involved in the down-regulation of TβRII through histone deacetylation. We have immunoprecipitated the protein complexes by biotinylated oligonucleotides corresponding to the HDI-responsive element in the TβRII promoter (-127/-75) and identified the proteins/factors using proteomics studies. The transcriptional repressor Meis1/2 is involved in repressing the TβRII promoter activity, possibly through its recruitment by Sp1 and NF-YA to the promoter. These results suggest a mechanism for the downregulation of TβRII in lung cancer and that TGF-β tumor suppressor functions may be restored by HDIs in lung cancer patients with the loss of TβRII expression.
Figures





References
-
- Nagatake M, Takagi Y, Osada H, Uchida K, Mitsudomi T, Saji S, Shimokata K, Takahashi T, Takahashi T. Somatic in vivo alterations of the DPC4 gene at 18q21 in human lung cancers. Cancer Res. 1996;56:2718–2720. - PubMed
-
- Uchida K, Nagatake M, Osada H, Yatabe Y, Kondo M, Mitsudomi T, Masuda A, Takahashi T, Takahashi T. Somatic in vivo alterations of the JV18-1 gene at 18q21 in human lung cancers. Cancer Res. 1996;56:5583–5585. - PubMed
-
- Osada H, Tatematsu Y, Masuda A, Saito T, Sugiyama M, Yanagisawa K, Takahashi T. Heterogeneous transforming growth factor (TGF)-β unresponsiveness and loss of TGF-β receptor type II expression caused by histone deacetylation in lung cancer cell lines. Cancer Res. 2001;61:8331–8339. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical