

Topological structure of the space of phenotypes: the case of RNA neutral networks
- PMID: 22028856
- PMCID: PMC3196570
- DOI: 10.1371/journal.pone.0026324
Topological structure of the space of phenotypes: the case of RNA neutral networks
Erratum in
- PLoS One. 2011;6(12). doi: 10.1371/annotation/b3e79f42-7316-4ff8-9762-514120463813 doi: 10.1371/annotation/b3e79f42-7316-4ff8-9762-514120463813
- PLoS One. 2011;6(12). doi: 10.1371/annotation/e1599064-95cc-47c9-bbcc-042202bf0423 doi: 10.1371/annotation/e1599064-95cc-47c9-bbcc-042202bf0423
- PLoS One. 2011;6(12). doi:10.1371/annotation/0a0bed45-e421-4f6a-8d7f-f48194a14516
Abstract
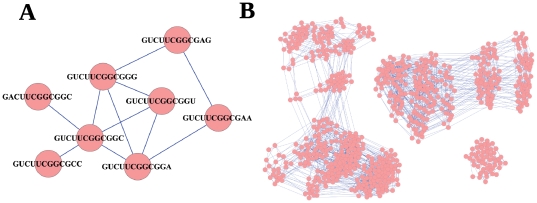
The evolution and adaptation of molecular populations is constrained by the diversity accessible through mutational processes. RNA is a paradigmatic example of biopolymer where genotype (sequence) and phenotype (approximated by the secondary structure fold) are identified in a single molecule. The extreme redundancy of the genotype-phenotype map leads to large ensembles of RNA sequences that fold into the same secondary structure and can be connected through single-point mutations. These ensembles define neutral networks of phenotypes in sequence space. Here we analyze the topological properties of neutral networks formed by 12-nucleotides RNA sequences, obtained through the exhaustive folding of sequence space. A total of 4(12) sequences fragments into 645 subnetworks that correspond to 57 different secondary structures. The topological analysis reveals that each subnetwork is far from being random: it has a degree distribution with a well-defined average and a small dispersion, a high clustering coefficient, and an average shortest path between nodes close to its minimum possible value, i.e. the Hamming distance between sequences. RNA neutral networks are assortative due to the correlation in the composition of neighboring sequences, a feature that together with the symmetries inherent to the folding process explains the existence of communities. Several topological relationships can be analytically derived attending to structural restrictions and generic properties of the folding process. The average degree of these phenotypic networks grows logarithmically with their size, such that abundant phenotypes have the additional advantage of being more robust to mutations. This property prevents fragmentation of neutral networks and thus enhances the navigability of sequence space. In summary, RNA neutral networks show unique topological properties, unknown to other networks previously described.
Conflict of interest statement
Figures


 as a function of the subnetwork size N. Colors correspond to one (black), two (red), three (green) and four (blue) base pairs in the secondary structure. The solid line corresponds to the numerical fitting
as a function of the subnetwork size N. Colors correspond to one (black), two (red), three (green) and four (blue) base pairs in the secondary structure. The solid line corresponds to the numerical fitting  (note the logarithmic-linear scale). The analytical approximation to
(note the logarithmic-linear scale). The analytical approximation to  making use of the values of
making use of the values of  ,
,  and α obtained from all the 12-nt folded sequences (and implying AS = 0.53) is plotted in long-dashed black line. The upper and lower bounds to coefficient AS yield
and α obtained from all the 12-nt folded sequences (and implying AS = 0.53) is plotted in long-dashed black line. The upper and lower bounds to coefficient AS yield  and
and  (plotted in short-dashed red lines).
(plotted in short-dashed red lines).
 , green stars). Circle colors correspond to the number of base pairs of each subnetwork (see caption of Fig. 3). In both plots (A) and (B), the analytical approximations using the values of
, green stars). Circle colors correspond to the number of base pairs of each subnetwork (see caption of Fig. 3). In both plots (A) and (B), the analytical approximations using the values of  ,
,  and α obtained from all the 12-nt folded sequences are plotted in long-dashed black lines.
and α obtained from all the 12-nt folded sequences are plotted in long-dashed black lines.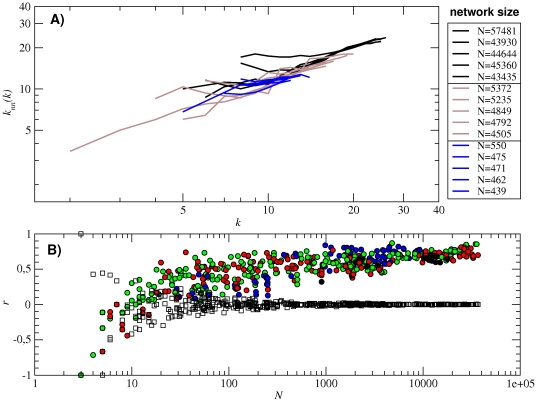

 , green stars). Circle colors correspond to the number of base pairs of each subnetwork (see caption of Fig. 3). The numerical fitting is plotted as a solid black line, while the analytical approximations correspond to the long-dashed black lines (for values of α and AS numerically obtained from the folding of all 12-nt sequences). Inset (A): relation between the average shortest path
, green stars). Circle colors correspond to the number of base pairs of each subnetwork (see caption of Fig. 3). The numerical fitting is plotted as a solid black line, while the analytical approximations correspond to the long-dashed black lines (for values of α and AS numerically obtained from the folding of all 12-nt sequences). Inset (A): relation between the average shortest path  and the average Hamming distance
and the average Hamming distance  of the subnetworks. Inset (B): relation between the longest distance between any pair of nodes of the network dmax and the maximum number of different bases between sequences Hmax (maximum Hamming distance). In the insets, the dashed lines are
of the subnetworks. Inset (B): relation between the longest distance between any pair of nodes of the network dmax and the maximum number of different bases between sequences Hmax (maximum Hamming distance). In the insets, the dashed lines are  and
and  , which correspond to the lower bounds of
, which correspond to the lower bounds of  and
and  , respectively.
, respectively.
 . Solid line in the inset is
. Solid line in the inset is  .
.

References
-
- Fontana W, Schuster P. Shaping space: The possible and the attainable in RNA genotypephenotype mapping. J Theor Biol. 1998;194:491–515. - PubMed
-
- Schuster P. Molecular insights into evolution of phenotypes. In: Crutchfield JP, Schuster P, editors. Evolutionary Dynamics. Oxford Univ. Press; 2003. pp. 163–215.
-
- Schuster P. Prediction of RNA secondary structures: from theory to models and real molecules. Rep Prog Phys. 2006;69:1419–1477.
-
- Grüner W, Giegerich R, Strothmann D, Reidys C, Weber J, et al. Analysis of RNA sequence structure maps by exhaustive enumeration. II. Structures of neutral networks and shape space covering. Monatsh Chem. 1996;127:375–389.
-
- Fontana W, Konings DAM, Stadler PF, Schuster P. Statistics of RNA secondary structures. Biopolymers. 1993;33:1389–1404. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
