

PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells
- PMID: 22031597
- PMCID: PMC3328839
- DOI: 10.1152/ajpcell.00337.2011
PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells
Abstract
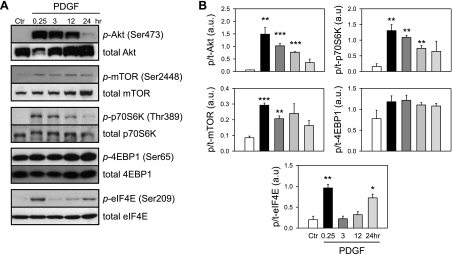
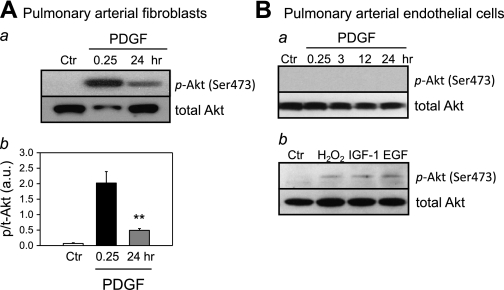
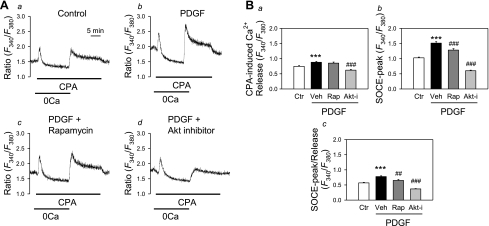
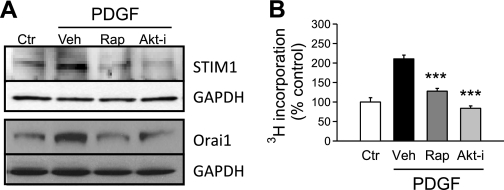
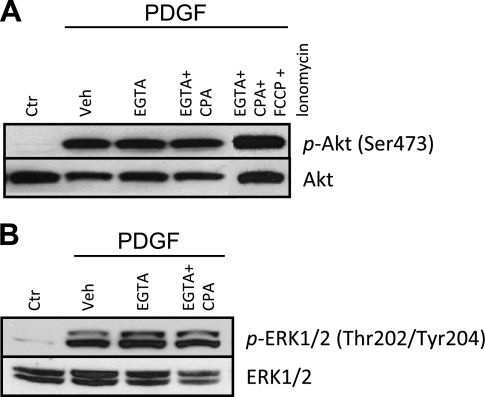
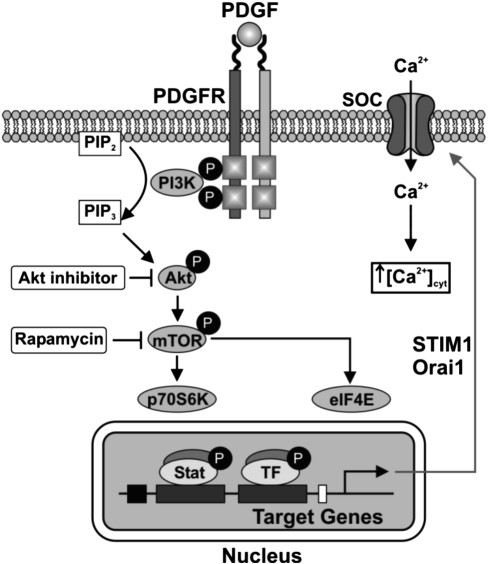
Platelet-derived growth factor (PDGF) and its receptor are known to be substantially elevated in lung tissues and pulmonary arterial smooth muscle cells (PASMC) isolated from patients and animals with pulmonary arterial hypertension. PDGF has been shown to phosphorylate and activate Akt and mammalian target of rapamycin (mTOR) in PASMC. In this study, we investigated the role of PDGF-mediated activation of Akt signaling in the regulation of cytosolic Ca(2+) concentration and cell proliferation. PDGF activated the Akt/mTOR pathway and, subsequently, enhanced store-operated Ca(2+) entry (SOCE) and cell proliferation in human PASMC. Inhibition of Akt attenuated the increase in cytosolic Ca(2+) concentration due to both SOCE and PASMC proliferation. This effect correlated with a significant downregulation of stromal interacting molecule (STIM) and Orai, proposed molecular correlates for SOCE in many cell types. The data from this study present a novel pathway for the regulation of Ca(2+) signaling and PASMC proliferation involving activation of Akt in response to upregulated expression of PDGF. Targeting this pathway may lead to the development of a novel therapeutic option for the treatment of pulmonary arterial hypertension.
Figures
References
-
- Balciunaite E, Jones S, Toker A, Kazlauskas A. PDGF initiates two distinct phases of protein kinase C activity that make unequal contributions to the G0 to S transition. Curr Biol 10: 261–267, 2000 - PubMed
-
- Bonnet S, Paulin R, Sutendra G, Dromparis P, Roy M, Watson KO, Nagendran J, Haromy A, Dyck JRB, Michelakis ED. Dehydroepiandrosterone reverses systemic vascular remodeling through the inhibition of the Akt/GSK3-β/NFAT axis. Circulation 120: 1231–1240, 2009 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous