

TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation
- PMID: 22033934
- PMCID: PMC3234870
- DOI: 10.1074/jbc.M111.310599
TLR2 and RIP2 pathways mediate autophagy of Listeria monocytogenes via extracellular signal-regulated kinase (ERK) activation
Abstract
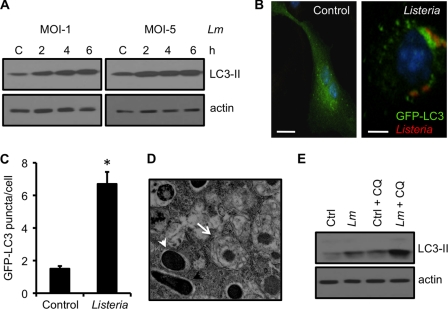
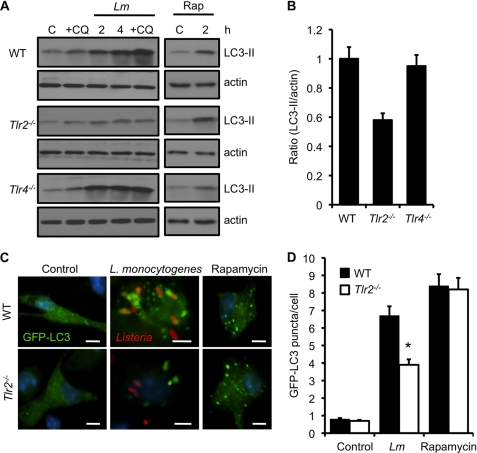
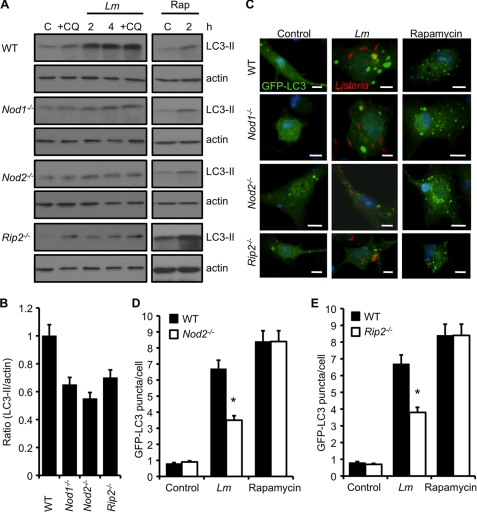
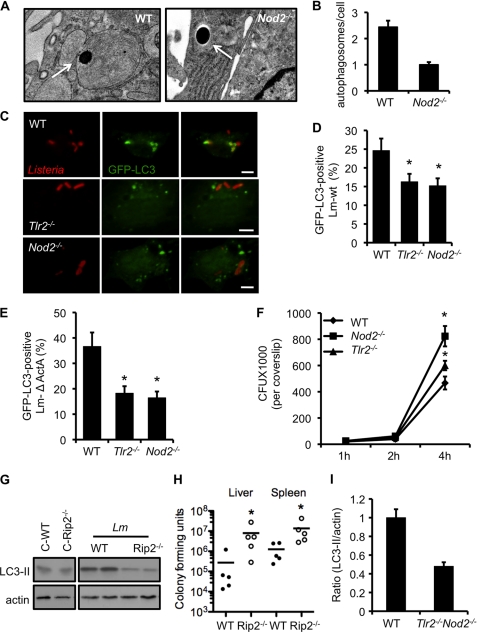
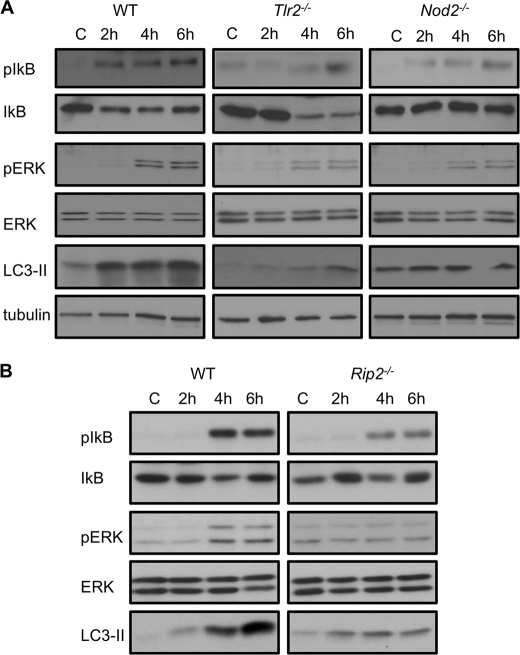
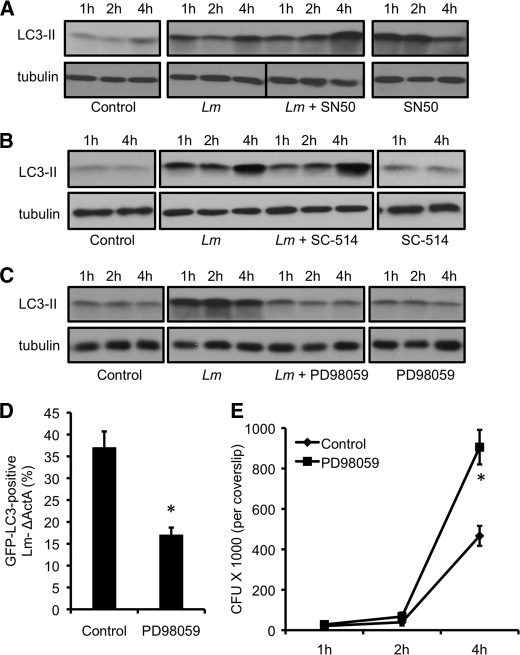
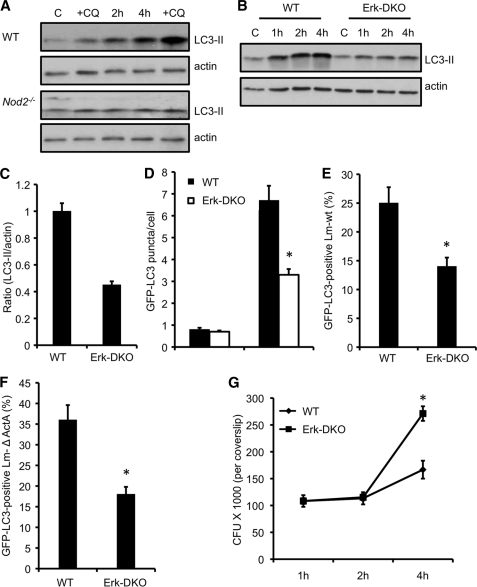
Listeria monocytogenes is a facultative intracellular pathogen that invades both phagocytic and non-phagocytic cells. Recent studies have shown that L. monocytogenes infection activates the autophagy pathway. However, the innate immune receptors involved and the downstream signaling pathways remain unknown. Here, we show that macrophages deficient in the TLR2 and NOD/RIP2 pathway display defective autophagy induction in response to L. monocytogenes. Inefficient autophagy in Tlr2(-/-) and Nod2(-/-) macrophages led to a defect in bacteria colocalization with the autophagosomal marker GFP-LC3. Consequently, macrophages lacking TLR2 and NOD2 were found to be more susceptible to L. monocytogenes infection, as were the Rip2(-/-) mice. Tlr2(-/-) and Nod2(-/-) cells showed perturbed NF-κB and ERK signaling. However, autophagy against L. monocytogenes was dependent selectively on the ERK pathway. In agreement, wild-type cells treated with a pharmacological inhibitor of ERK or ERK-deficient cells displayed inefficient autophagy activation in response to L. monocytogenes. Accordingly, fewer bacteria were targeted to the autophagosomes and, consequently, higher bacterial growth was observed in cells deficient in the ERK signaling pathway. These findings thus demonstrate that TLR2 and NOD proteins, acting via the downstream ERK pathway, are crucial to autophagy activation and provide a mechanistic link between innate immune receptors and induction of autophagy against cytoplasm-invading microbes, such as L. monocytogenes.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
