

Mutations in multiple PKD genes may explain early and severe polycystic kidney disease
- PMID: 22034641
- PMCID: PMC3279997
- DOI: 10.1681/ASN.2010101080
Mutations in multiple PKD genes may explain early and severe polycystic kidney disease
Abstract
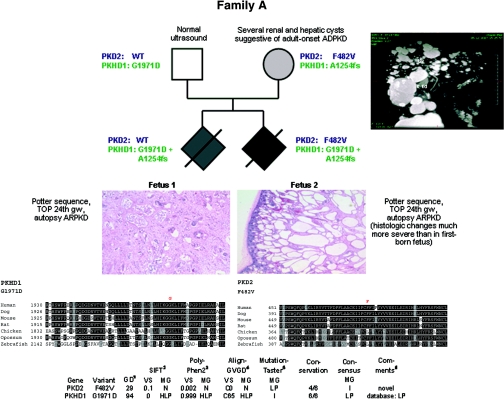
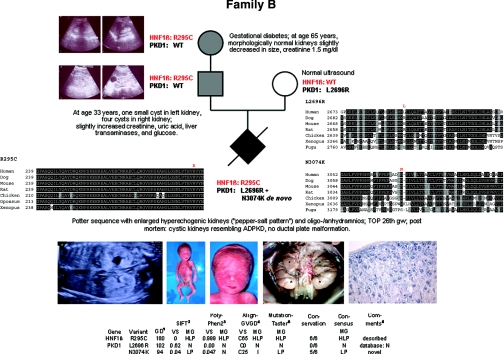
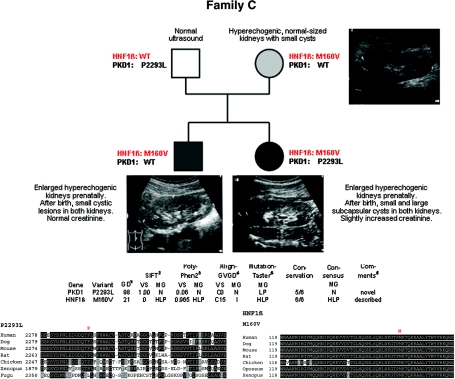
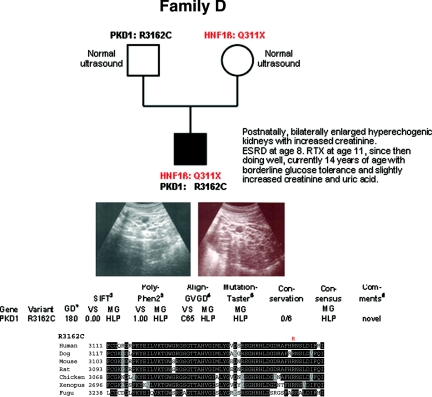
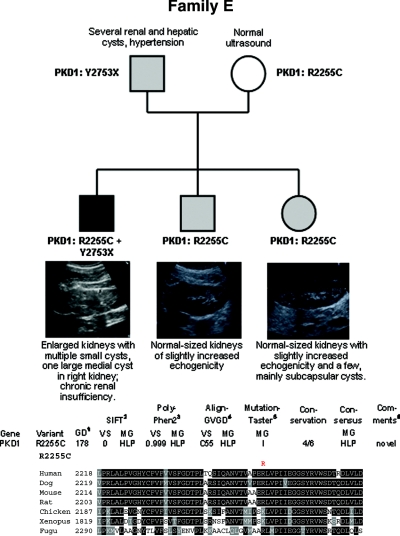
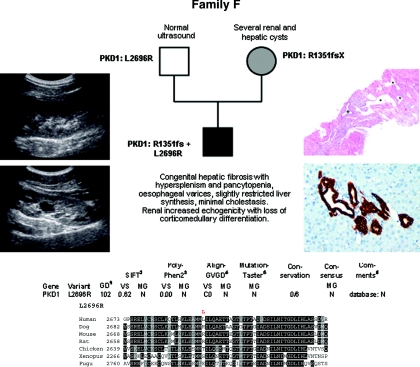
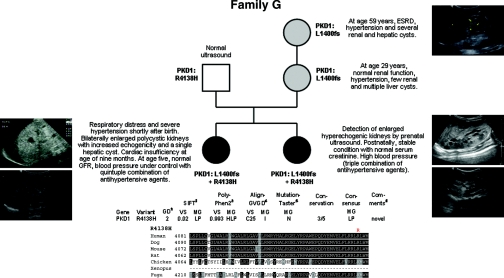
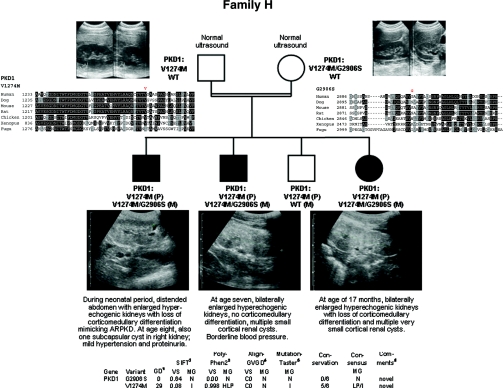
Autosomal dominant polycystic kidney disease (ADPKD) is typically a late-onset disease caused by mutations in PKD1 or PKD2, but about 2% of patients with ADPKD show an early and severe phenotype that can be clinically indistinguishable from autosomal recessive polycystic kidney disease (ARPKD). The high recurrence risk in pedigrees with early and severe PKD strongly suggests a common familial modifying background, but the mechanisms underlying the extensive phenotypic variability observed among affected family members remain unknown. Here, we describe severely affected patients with PKD who carry, in addition to their expected familial germ-line defect, additional mutations in PKD genes, including HNF-1β, which likely aggravate the phenotype. Our findings are consistent with a common pathogenesis and dosage theory for PKD and may propose a general concept for the modification of disease expression in other so-called monogenic disorders.
Figures
References
-
- Wilson PD: Polycystic kidney disease. N Engl J Med 350: 151–164, 2004 - PubMed
-
- Torres VE, Harris PC, Pirson Y: Autosomal dominant polycystic kidney disease. Lancet 369: 1287–1301, 2007 - PubMed
-
- Bergmann C, Zerres K: Early manifestations of polycystic kidney disease. Lancet 369: 2157, 2007 - PubMed
-
- Ogborn MR: Polycystic kidney disease: A truly pediatric problem. Pediatr Nephrol 8: 762–767, 1994 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
