

Molecular dynamics simulations and drug discovery
- PMID: 22035460
- PMCID: PMC3203851
- DOI: 10.1186/1741-7007-9-71
Molecular dynamics simulations and drug discovery
Abstract
This review discusses the many roles atomistic computer simulations of macromolecular (for example, protein) receptors and their associated small-molecule ligands can play in drug discovery, including the identification of cryptic or allosteric binding sites, the enhancement of traditional virtual-screening methodologies, and the direct prediction of small-molecule binding energies. The limitations of current simulation methodologies, including the high computational costs and approximations of molecular forces required, are also discussed. With constant improvements in both computer power and algorithm design, the future of computer-aided drug design is promising; molecular dynamics simulations are likely to play an increasingly important role.
Figures
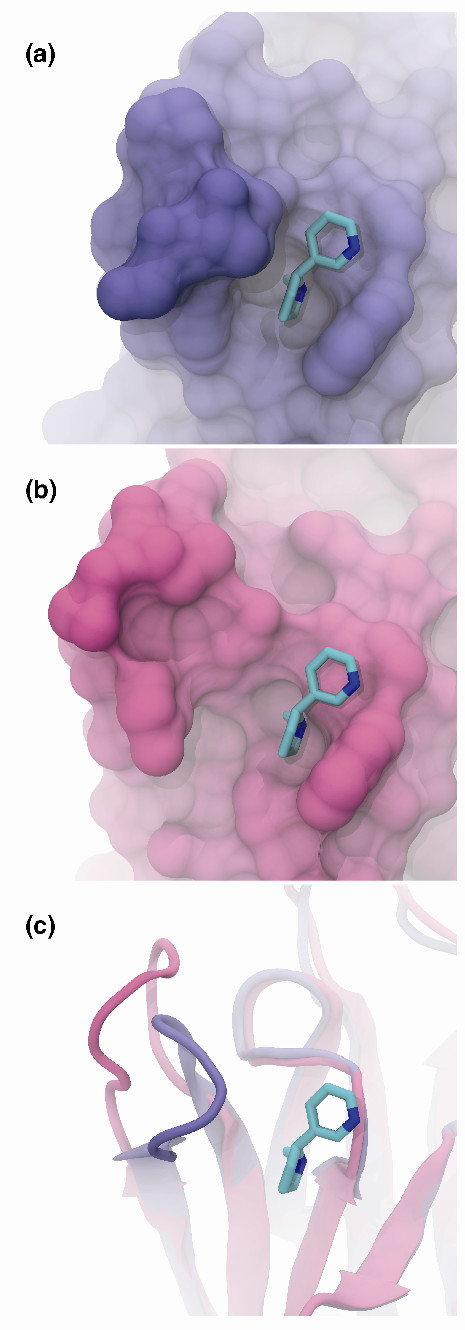
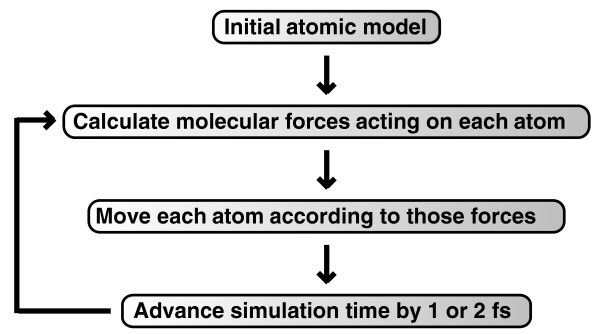
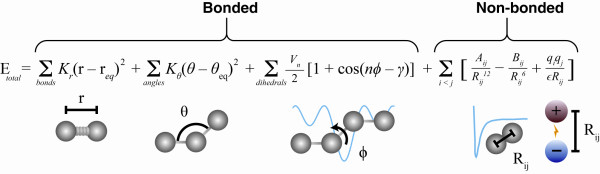
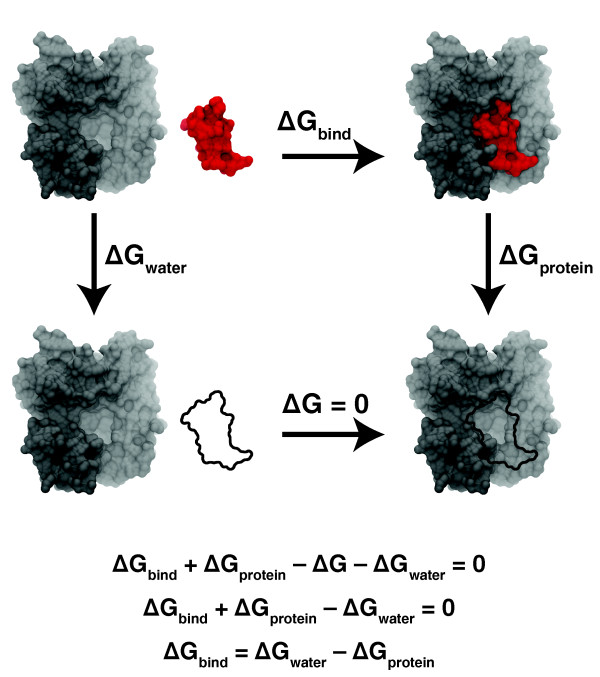
References
-
- Feynman RP. QED: the Strange Theory of Light and Matter. Princeton, NJ: Princeton University Press; 1985.
-
- Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber Dtsch Chem Ges. 1894;27:2985–2993. doi: 10.1002/cber.18940270364. - DOI
