

doi: 10.1016/j.cell.2011.10.011.
The seeds of neurodegeneration: prion-like spreading in ALS
Affiliations
- PMID: 22036560
- PMCID: PMC3220614
- DOI: 10.1016/j.cell.2011.10.011
Item in Clipboard
The seeds of neurodegeneration: prion-like spreading in ALS
Cell.
.
Abstract
Misfolded proteins accumulating in several neurodegenerative diseases (including Alzheimer, Parkinson, and Huntington diseases) can cause aggregation of their native counterparts through a mechanism similar to the infectious prion protein's induction of a pathogenic conformation onto its cellular isoform. Evidence for such a prion-like mechanism has now spread to the main misfolded proteins, SOD1 and TDP-43, implicated in amyotrophic lateral sclerosis (ALS). The major neurodegenerative diseases may therefore have mechanistic parallels for non-cell-autonomous spread of disease within the nervous system.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
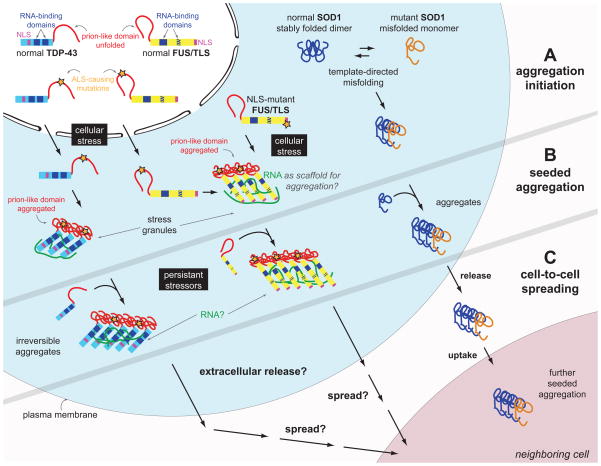
A. Mutant, misfolded SOD1 was shown to induce the misfolding of its native counterpart, in a template-directed reaction, thereby forming a seed of aggregated protein. TDP-43 and FUS/TLS are both incorporated in stress granules, which form through the ordered aggregation of several RNA-binding proteins complexed with RNA molecules. This physiologic reaction to cellular stress may be the initial trigger for pathogenic inclusion formation since the increased local protein concentration and RNA scaffolding molecules may facilitate ordered aggregation of TDP-43 and/or FUS/TLS. Mutations in the prion-like domain of TDP-43 (and maybe also FUS/TLS) enhance its aggregation propensity, while mutations in the nuclear localization domain (NLS) of FUS/TLS increase its cytoplasmic localization. B. Misfolded SOD1 follows a self-perpetuating seeding reaction in cell culture. Upon chronic cellular stress and defects in stress granule disassembly occurring with aging, the functional prion-like conformational changes of TDP-43 and FUS/TLS associated with their physiological roles in stress granule formation may transform into pathogenic self-perpetuating, irreversible aggregation. It is unknown whether cellular RNA is occasionally trapped within the cytoplasmic FUS/TLS and/or TDP-43 inclusions, thereby depleting the cell of essential RNA components. C. SOD1 aggregates transfer from cell-to-cell to initiate misfolding and aggregation of native SOD1 in neighboring cells (shown in cell culture). It is currently not known whether TDP-43 and/or FUS/TLS can spread from cell to cell by a similar mechanism. Filled blue boxes on TDP-43 and FUS/TLS molecules indicate RNA-recognition motifs and the striped blue box on FUS/TLS refers to the zinc finger domain that can also bind RNA.
References
-
- Aguzzi A. Cell biology: Beyond the prion principle. Nature. 2009;459:924–925. - PubMed
-
- Aguzzi A, Rajendran L. The transcellular spread of cytosolic amyloids, prions, and prionoids. Neuron. 2009;64:783–790. - PubMed
-
- Aguzzi A, Sigurdson C, Heikenwaelder M. Molecular mechanisms of prion pathogenesis. Annu Rev Pathol. 2008;3:11–40. - PubMed
-
- Anderson P, Kedersha N. RNA granules: post-transcriptional and epigenetic modulators of gene expression. Nat Rev Mol Cell Biol. 2009;10:430–436. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
