

The distribution of immunomodulatory cells in the lungs of patients with idiopathic pulmonary fibrosis
- PMID: 22037258
- PMCID: PMC3270219
- DOI: 10.1038/modpathol.2011.166
The distribution of immunomodulatory cells in the lungs of patients with idiopathic pulmonary fibrosis
Abstract
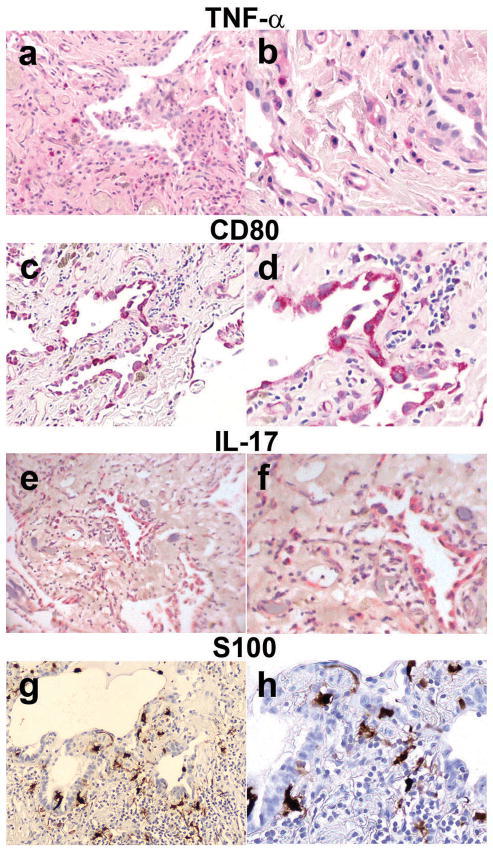
We have characterized the immune system involvement in the disease processes of idiopathic pulmonary fibrosis in novel ways. To do so, we analyzed lung tissue from 21 cases of idiopathic pulmonary fibrosis and 21 (non-fibrotic, non-cancerous) controls for immune cell and inflammation-related markers. The immunohistochemical analysis of the tissue was grouped by patterns of severity in disease pathology. There were significantly greater numbers of CD68(+) and CD80(+) cells and significantly fewer CD3(+), CD4(+), and CD45RO(+) cells in areas of relatively (histologically) normal lung in biopsy samples from idiopathic pulmonary fibrosis patients compared with controls. In zones of active disease, characterized by epithelial cell regeneration and fibrosis, there were significantly more cells expressing CD4, CD8, CD20, CD68, CD80, chemokine receptor 6 (CCR6), S100, IL-17, tumor necrosis factor-α, and retinoic acid-related orphan receptors compared with histologically normal lung areas from idiopathic pulmonary fibrosis patients. Inflammation was implicated in these active regions by the cells that expressed retinoid orphan receptor-α, -β, and -γ, CCR6, and IL-17. The regenerating epithelial cells predominantly expressed these pro-inflammatory molecules, as evidenced by co-expression analyses with epithelial cytokeratins. Macrophages in pseudo-alveoli and CD3(+) T cells in the fibrotic interstitium also expressed IL-17. Co-expression of IL-17 with retinoid orphan receptors and epithelial cytoskeletal proteins, CD68, and CD3 in epithelial cells, macrophages, and T-cells, respectively, confirmed the production of IL-17 by these cell types. There was little staining for forkhead box p3, CD56, or CD34 in any idiopathic pulmonary fibrosis lung regions. The fibrotic regions had fewer immune cells overall. In summary, our study shows participation of innate and adaptive mononuclear cells in active-disease regions of idiopathic pulmonary fibrosis lung, where the regenerating epithelial cells appear to propagate inflammation. The regenerative mechanisms become skewed to ultimately result in lethal, fibrotic restriction of lung function.
Conflict of interest statement
The authors have no conflicts of interest to disclose.
Figures








References
-
- Olsen AL, Swigris JJ, Lezotte DC, Norris JM, Wilson CG, Brown KK. Mortality from pulmonary fibrosis increased in the United States from 1992 to 2003. Am J Respir Crit Care Med. 2007;176:277–84. - PubMed
-
- Katzenstein A-LA, Mukhopadhyay S, Myers JL. Diagnosis of usual interstitial pneumonia and distinction from other fibrosing interstitial lung diseases. Hum Pathol. 2008;39:1562–81. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
