

Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer's disease: implications to mitochondria-targeted antioxidant therapeutics
- PMID: 22037588
- PMCID: PMC3272314
- DOI: 10.1016/j.bbadis.2011.10.011
Abnormal mitochondrial dynamics and synaptic degeneration as early events in Alzheimer's disease: implications to mitochondria-targeted antioxidant therapeutics
Abstract
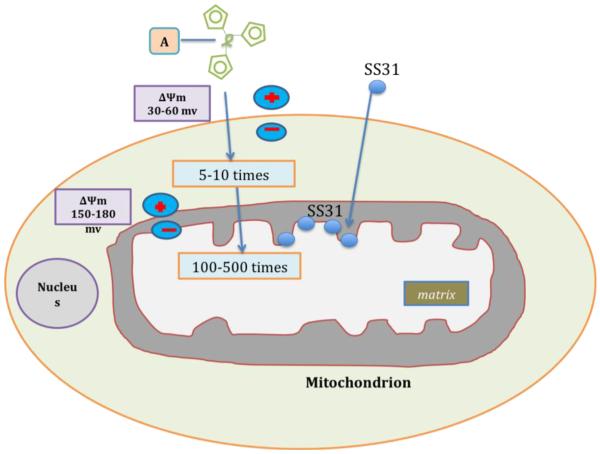
Synaptic pathology and mitochondrial oxidative damage are early events in Alzheimer's disease (AD) progression. Loss of synapses and synaptic damage are the best correlates of cognitive deficits found in AD patients. Recent research on amyloid beta (Aβ) and mitochondria in AD revealed that Aβ accumulates in synapses and synaptic mitochondria, leading to abnormal mitochondrial dynamics and synaptic degeneration in AD neurons. Further, recent studies using live-cell imaging and primary neurons from amyloid beta precursor protein (AβPP) transgenic mice revealed reduced mitochondrial mass, defective axonal transport of mitochondria and synaptic degeneration, indicating that Aβ is responsible for mitochondrial and synaptic deficiencies. Tremendous progress has been made in studying antioxidant approaches in mouse models of AD and clinical trials of AD patients. This article highlights the recent developments made in Aβ-induced abnormal mitochondrial dynamics, defective mitochondrial biogenesis, impaired axonal transport and synaptic deficiencies in AD. This article also focuses on mitochondrial approaches in treating AD, and also discusses latest research on mitochondria-targeted antioxidants in AD. This article is part of a Special Issue entitled: Antioxidants and Antioxidant Treatment in Disease.
Copyright © 2011 Elsevier B.V. All rights reserved.
Figures
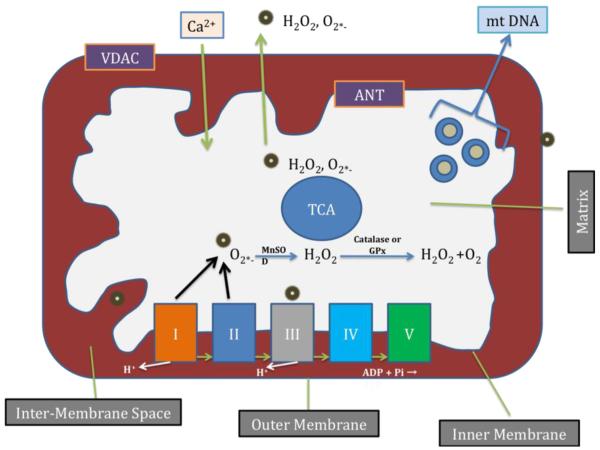
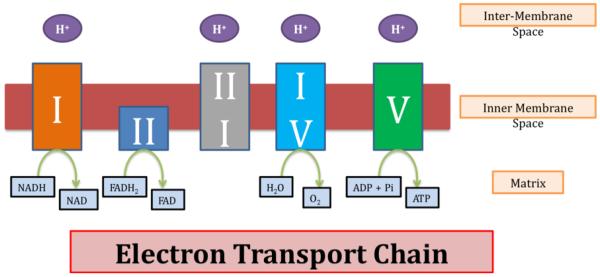
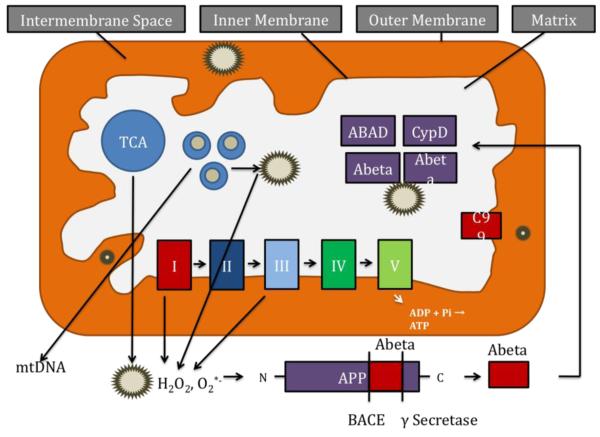
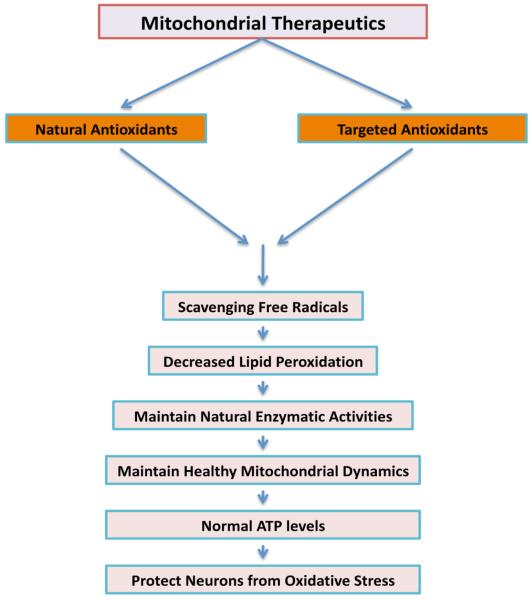
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
