

RIP1-mediated regulation of lymphocyte survival and death responses
- PMID: 22038529
- PMCID: PMC3244575
- DOI: 10.1007/s12026-011-8249-3
RIP1-mediated regulation of lymphocyte survival and death responses
Abstract
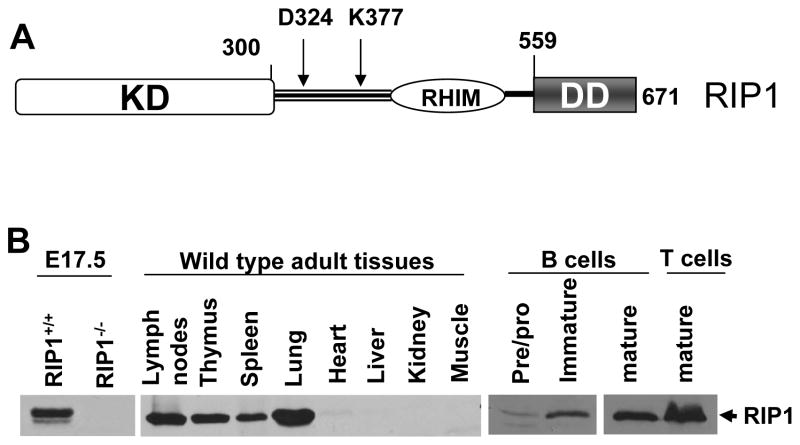
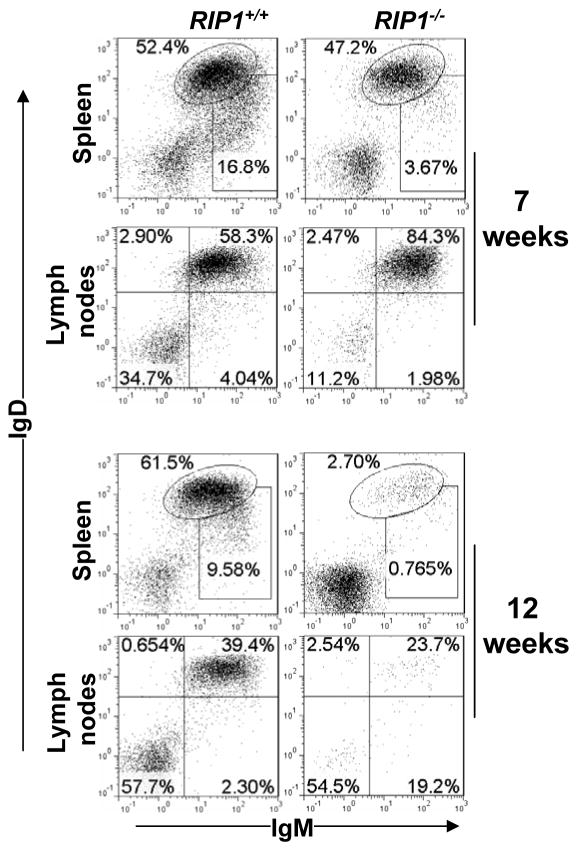
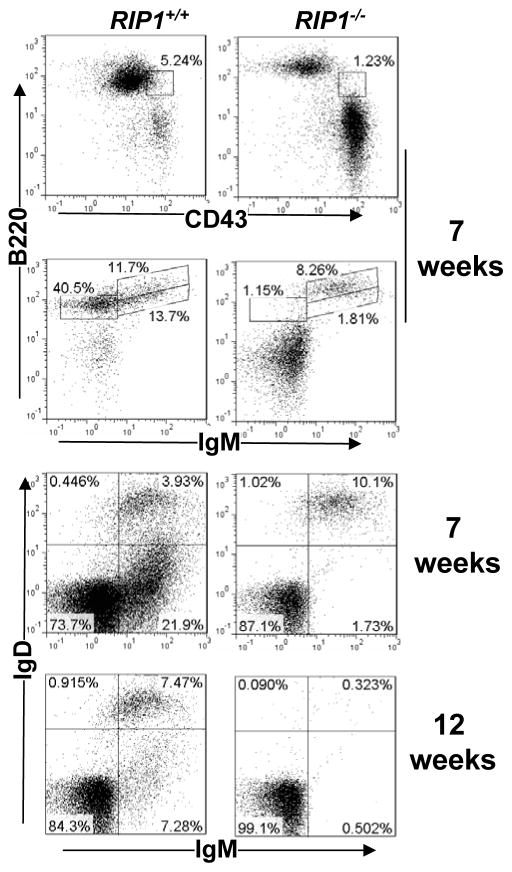
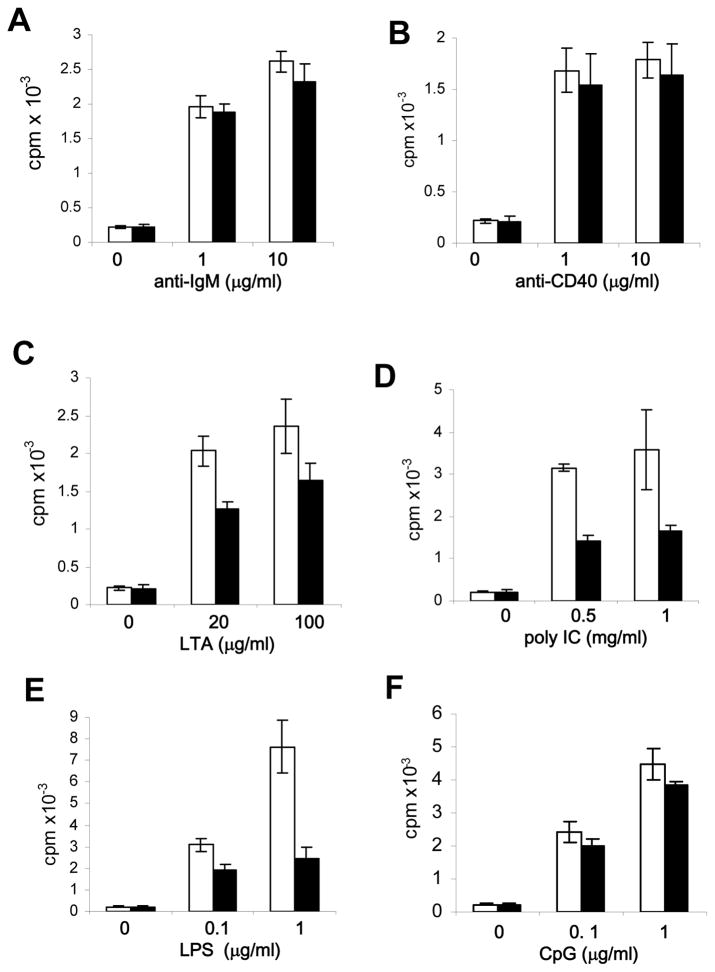
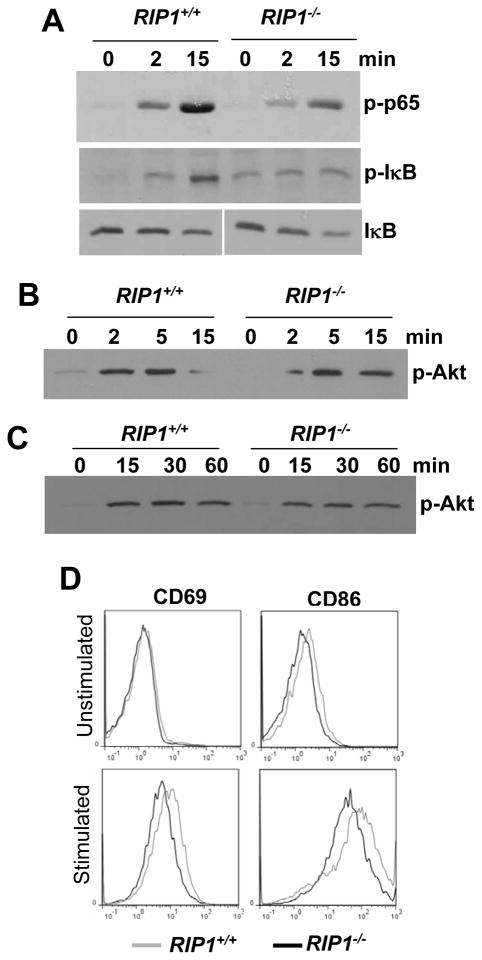
RIP1 is an adaptor serine/threonine kinase associated with the signaling complex of death receptors (DRs) including Fas, TNFR1, and TRAIL-Rs which can initiate apoptosis. While DRs are dispensable throughout development, RIP1 deletion results in perinatal lethality. The developmental defect caused by absence of RIP1 remains unexplained. In previous studies, RIP1-deficient hematopoietic progenitors failed to reconstitute the T cell compartment and our recent data indicate a new role for RIP1 in TCR-induced activation of the pro-survival NF-κB pathway. Here, we show that RIP1 is also critical for B cell development. In addition, RIP1(-/-) B cells stimulated through LPS/TLR4 are impaired in NF-κB activation but have no major defect in the Akt pathway. Recently, RIP1 has also emerged as a critical player in necrosis-like death, necroptosis, in various cell lines. We have demonstrated that RIP1 deficiency can reverse the embryonic and T cell proliferation defects in mice lacking FADD, a caspase adaptor protein, which indicates a potential role for RIP1 in mediating in vivo necroptosis. We provide an overview and discussion of the accumulating data revealing insights into the diverse functions of RIP1 in survival and death signaling in lymphocytes.
Figures
References
-
- Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–1455. - PubMed
-
- Locksley RM, Killeen N, Lenardo MJ. The TNF and TNF Receptor Superfamilies. Integrating Mammalian Biology Cell. 2001;104:487–501. - PubMed
-
- Wilson NS, Dixit V, Ashkenazi A. Death receptor signal transducers: nodes of coordination in immune signaling networks. Nat Immunol. 2009;10:348–355. - PubMed
-
- Ashkenazi A, Dixit VM. Apoptosis control by death and decoy receptors. Curr Opin Cell Biol. 1999;11:255–260. - PubMed
-
- Itoh N, Nagata S. A novel protein domain required for apoptosis. J Biol Chem. 1993;268:10932–10937. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
