

Calibration of DFT Functionals for the Prediction of Fe Mössbauer Spectral Parameters in Iron-Nitrosyl and Iron-Sulfur Complexes: Accurate Geometries Prove Essential
- PMID: 22039359
- PMCID: PMC3203024
- DOI: 10.1021/ct200187d
Calibration of DFT Functionals for the Prediction of Fe Mössbauer Spectral Parameters in Iron-Nitrosyl and Iron-Sulfur Complexes: Accurate Geometries Prove Essential
Abstract
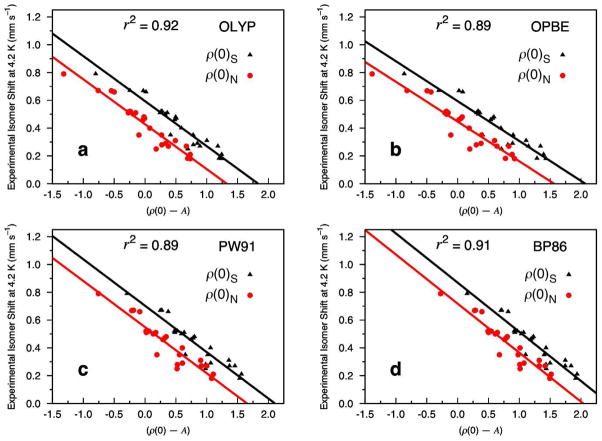
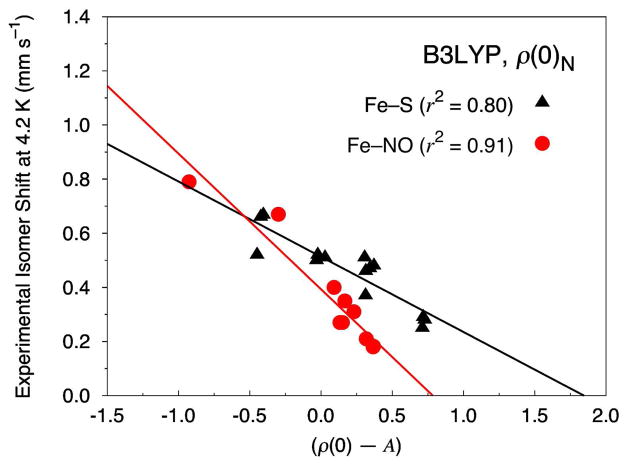
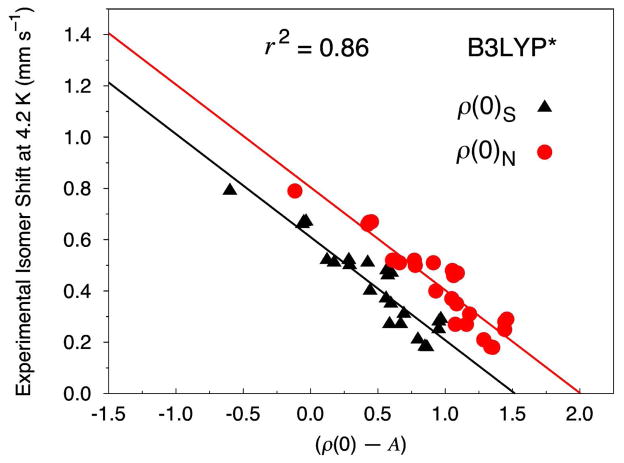
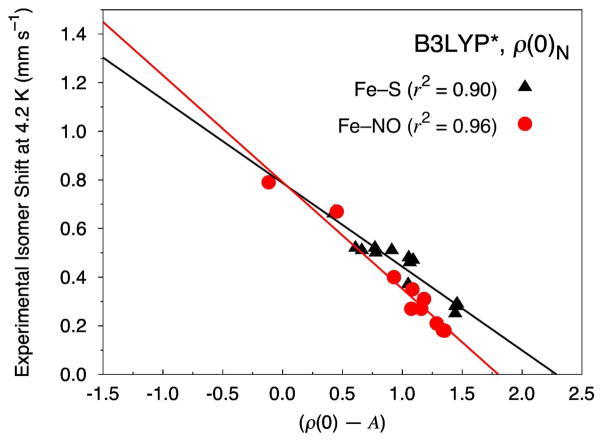
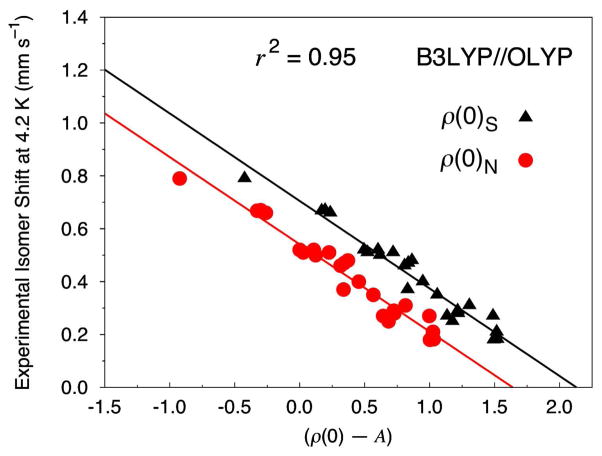
Six popular density functionals in conjunction with the conductor-like screening (COSMO) solvation model have been used to obtain linear Mössbauer isomer shift (IS) and quadrupole splitting (QS) parameters for a test set of 20 complexes (with 24 sites) comprised of nonheme nitrosyls (Fe-NO) and non-nitrosyl (Fe-S) complexes. For the first time in an IS analysis, the Fe electron density was calculated both directly at the nucleus, ρ(0)(N), which is the typical procedure, and on a small sphere surrounding the nucleus, ρ(0)(S), which is the new standard algorithm implemented in the ADF software package. We find that both methods yield (near) identical slopes from each linear regression analysis but are shifted with respect to ρ(0) along the x-axis. Therefore, the calculation of the Fe electron density with either method gives calibration fits with equal predictive value. Calibration parameters obtained from the complete test set for OLYP, OPBE, PW91, and BP86 yield correlation coefficients (r(2)) of approximately 0.90, indicating that the calibration fit is of good quality. However, fits obtained from B3LYP and B3LYP* with both Slater-type and Gaussian-type orbitals are generally found to be of poorer quality. For several of the complexes examined in this study, we find that B3LYP and B3LYP* give geometries that possess significantly larger deviations from the experimental structures than OLYP, OPBE, PW91 or BP86. This phenomenon is particularly true for the di- and tetranuclear Fe complexes examined in this study. Previous Mössbauer calibration fit studies using these functionals have usually included mononuclear Fe complexes alone, where these discrepancies are less pronounced. An examination of spin expectation values reveals B3LYP and B3LYP* approach the weak-coupling limit more closely than the GGA exchange-correlation functionals. The high degree of variability in our calculated S(2) values for the Fe-NO complexes highlights their challenging electronic structure. Significant improvements to the isomer shift calibrations are obtained for B3LYP and B3LYP* when geometries obtained with the OLYP functional are used. In addition, greatly improved performance of these functionals is found if the complete test set is grouped separately into Fe-NO and Fe-S complexes. Calibration fits including only Fe-NO complexes are found to be excellent, while those containing the non-nitrosyl Fe-S complexes alone are found to demonstrate less accurate correlations. Similar trends are also found with OLYP, OPBE, PW91, and BP86. Correlations between experimental and calculated QSs were also investigated. Generally, universal and separate Fe-NO and Fe-S fit parameters obtained to determine QSs are found to be of good to excellent quality for every density functional examined, especially if [Fe(4)(NO)(4)(μ(3)-S)(4)](-) is removed from the test set.
Figures
References
-
- Schünemann V, Paulsen H. Mössbauer Spectroscopy. In: Scott RA, Lukehart CM, editors. Applications of Physical Methods to Inorganic and Bioinorganic Chemistry. John Wiley & Sons Ltd; Chichester: 2007. pp. 243–69.
-
- Krebs C, Price JC, Baldwin J, Saleh L, Green MT, Bollinger JM. Inorg Chem. 2005;44:742–57. - PubMed
-
- Neese F. Inorg Chim Acta. 2002;337:181–92.
-
- Neese F. Coord Chem Rev. 2009;253:526–63.
-
- Siegbahn PEM. J Comput Chem. 2001;22:1634–45.
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous