

Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells
- PMID: 22041887
- PMCID: PMC3280908
- DOI: 10.4161/cbt.12.10.17780
Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells
Abstract
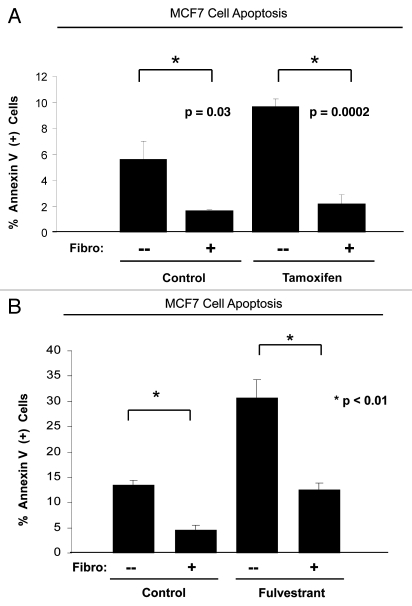
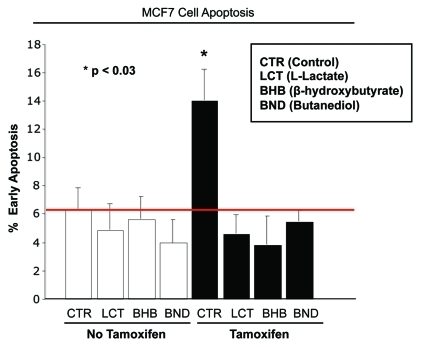
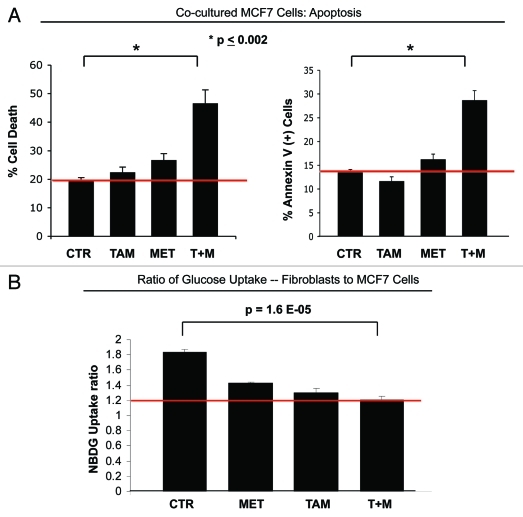
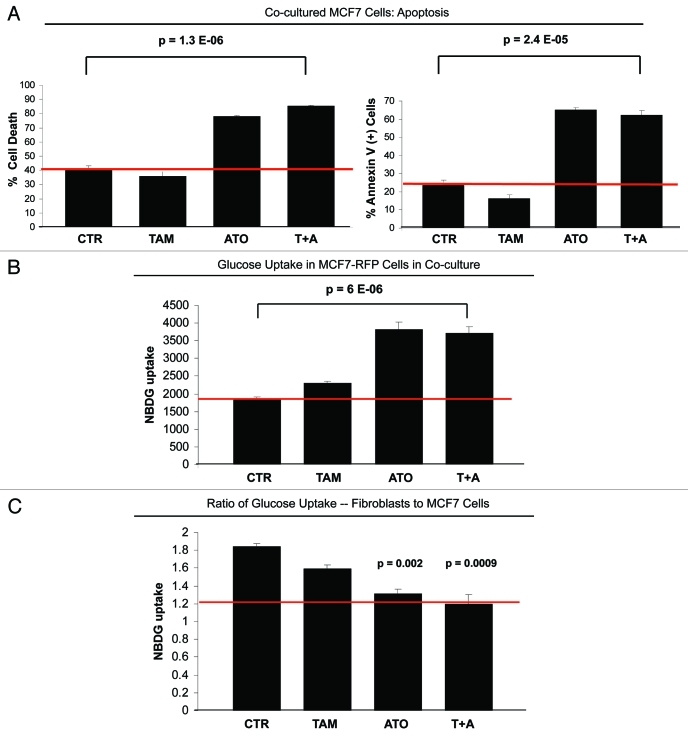
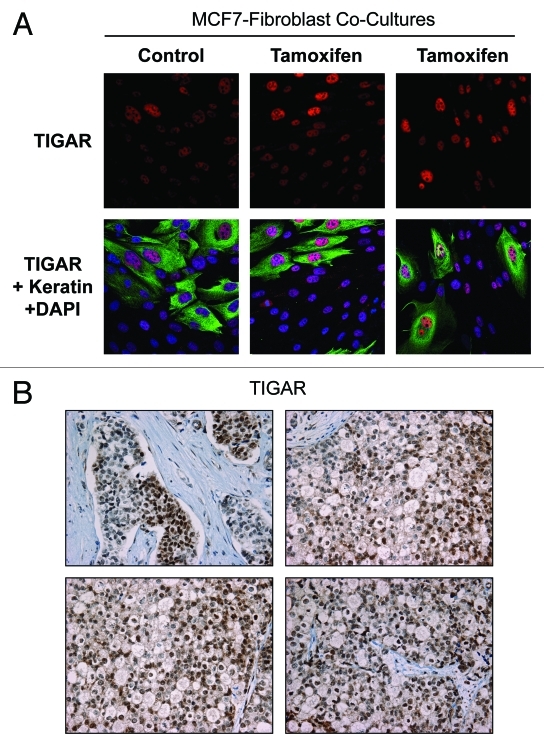
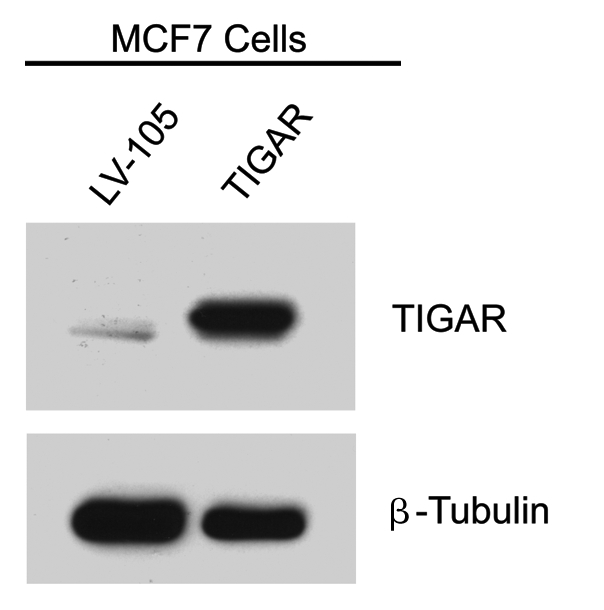
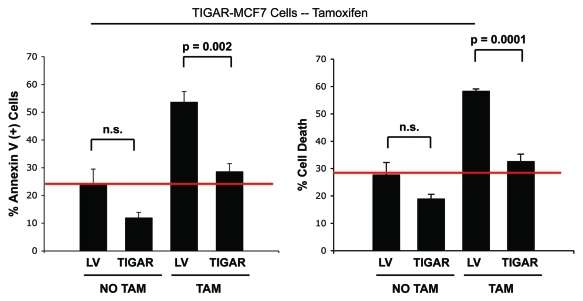
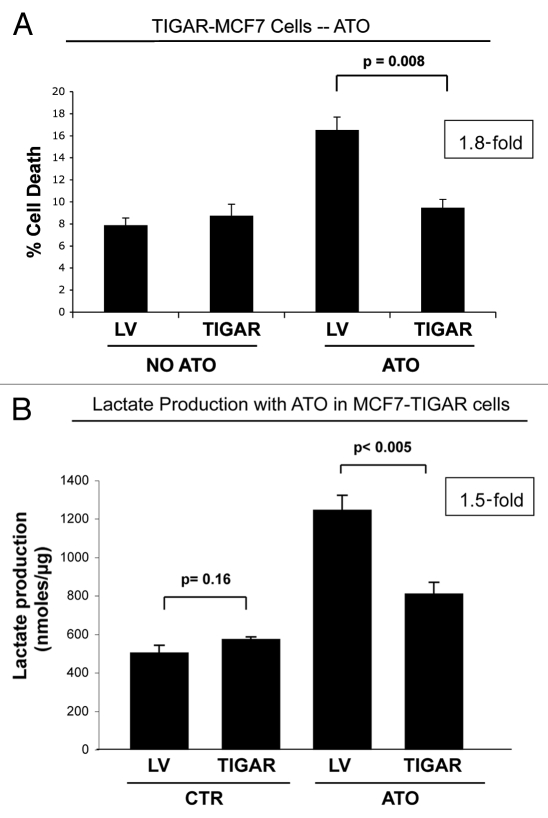
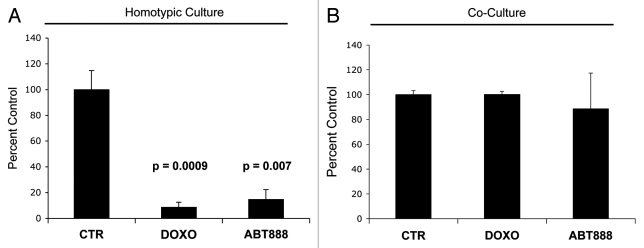
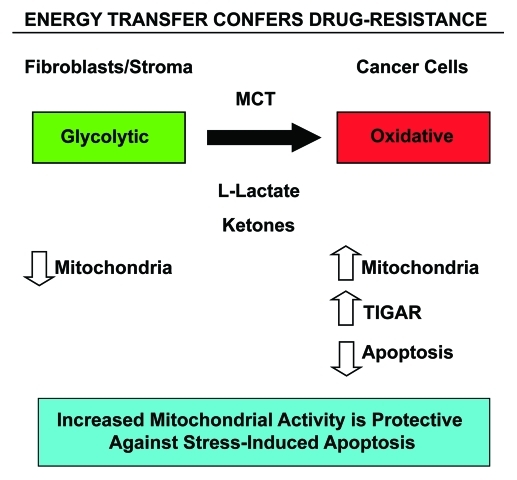
Here, we show that tamoxifen resistance is induced by cancer-associated fibroblasts (CAFs). Coculture of estrogen receptor positive (ER+) MCF7 cells with fibroblasts induces tamoxifen and fulvestrant resistance with 4.4 and 2.5-fold reductions, respectively, in apoptosis compared with homotypic MCF7 cell cultures. Treatment of MCF7 cells cultured alone with high-energy mitochondrial "fuels" (L-lactate or ketone bodies) is sufficient to confer tamoxifen resistance, mimicking the effects of coculture with fibroblasts. To further demonstrate that epithelial cancer cell mitochondrial activity is the origin of tamoxifen resistance, we employed complementary pharmacological and genetic approaches. First, we studied the effects of two mitochondrial "poisons," namely metformin and arsenic trioxide (ATO), on fibroblast-induced tamoxifen resistance. We show here that treatment with metformin or ATO overcomes fibroblast-induced tamoxifen resistance in MCF7 cells. Treatment with the combination of tamoxifen plus metformin or ATO leads to increases in glucose uptake in MCF7 cells, reflecting metabolic uncoupling between epithelial cancer cells and fibroblasts. In coculture, tamoxifen induces the upregulation of TIGAR (TP53-induced glycolysis and apoptosis regulator), a p53 regulated gene that simultaneously inhibits glycolysis, autophagy and apoptosis and reduces ROS generation, thereby promoting oxidative mitochondrial metabolism. To genetically mimic the effects of coculture, we next recombinantly overexpressed TIGAR in MCF7 cells. Remarkably, TIGAR overexpression protects epithelial cancer cells from tamoxifen-induced apoptosis, providing genetic evidence that increased mitochondrial function confers tamoxifen resistance. Finally, CAFs also protect MCF7 cells against apoptosis induced by other anticancer agents, such as the topoisomerase inhibitor doxorubicin (adriamycin) and the PARP-1 inhibitor ABT-888. These results suggest that the tumor microenvironment may be a general mechanism for conferring drug resistance. In summary, we have discovered that mitochondrial activity in epithelial cancer cells drives tamoxifen resistance in breast cancer and that mitochondrial "poisons" are able to re-sensitize these cancer cells to tamoxifen. In this context, TIGAR may be a key "druggable" target for preventing drug resistance in cancer cells, as it protects cancer cells against the onset of stress-induced mitochondrial dys-function and aerobic glycolysis.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
- R01 CA075503/CA/NCI NIH HHS/United States
- R01 CA098779/CA/NCI NIH HHS/United States
- R01-CA-120876/CA/NCI NIH HHS/United States
- R01 CA120876/CA/NCI NIH HHS/United States
- R01-CA-70896/CA/NCI NIH HHS/United States
- R01-CA-098779/CA/NCI NIH HHS/United States
- R01-CA-86072/CA/NCI NIH HHS/United States
- R01-AR-055660/AR/NIAMS NIH HHS/United States
- R01-CA-080250/CA/NCI NIH HHS/United States
- R01 CA070896/CA/NCI NIH HHS/United States
- R01 CA107382/CA/NCI NIH HHS/United States
- P30 CA056036/CA/NCI NIH HHS/United States
- P30-CA-56036/CA/NCI NIH HHS/United States
- R01-CA-107382/CA/NCI NIH HHS/United States
- R01 AR055660/AR/NIAMS NIH HHS/United States
- R01-CA-75503/CA/NCI NIH HHS/United States
- R01 CA080250/CA/NCI NIH HHS/United States
- R01 CA086072/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous