

Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4
- PMID: 22042619
- PMCID: PMC3206346
- DOI: 10.1083/jcb.201105063
Selective repression of MEF2 activity by PKA-dependent proteolysis of HDAC4
Abstract
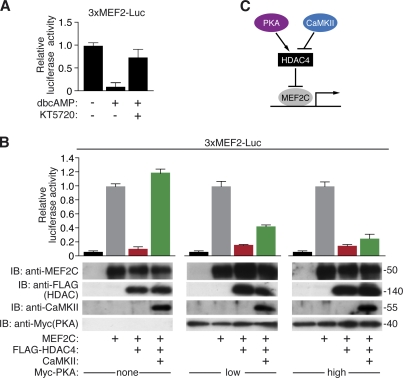
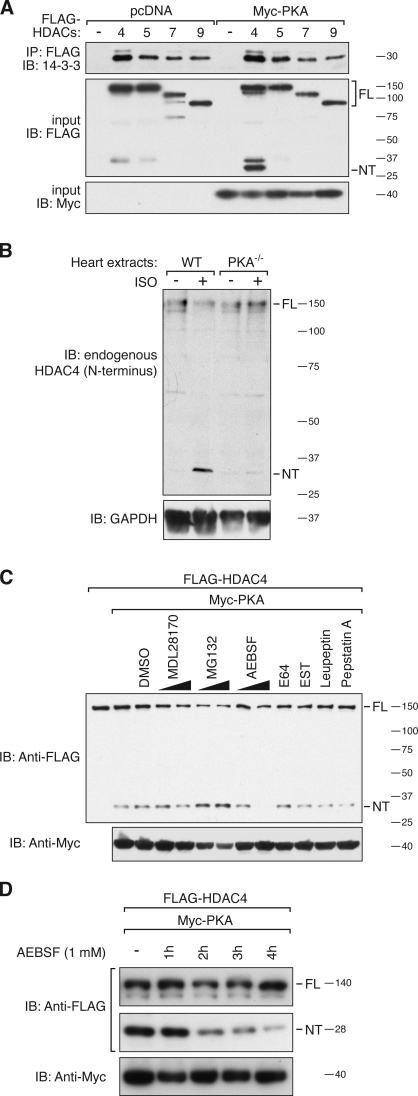
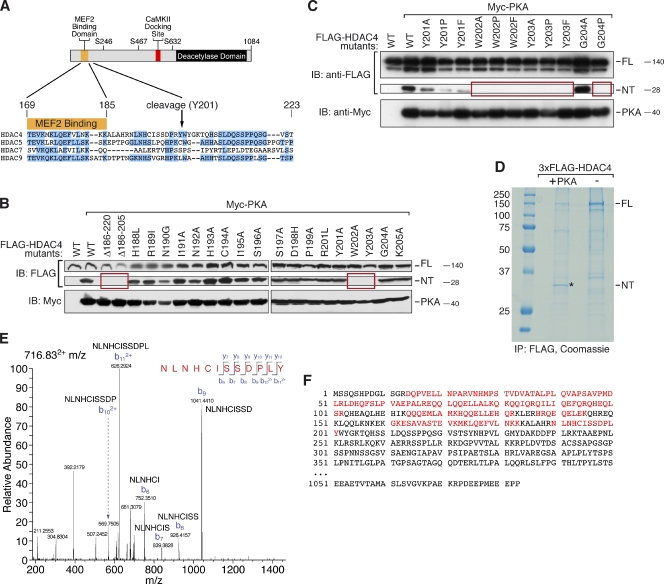
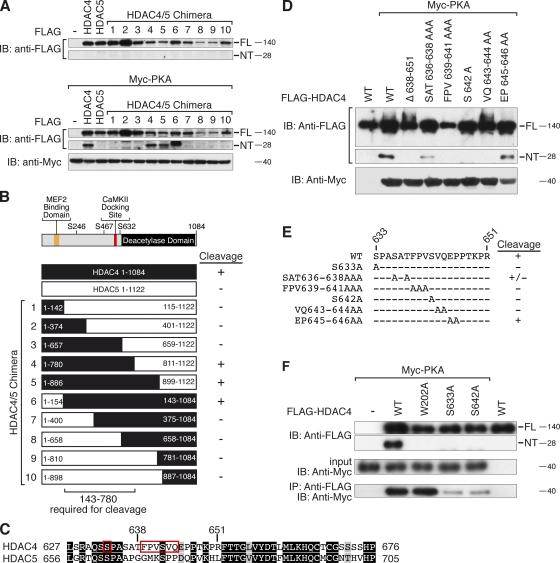
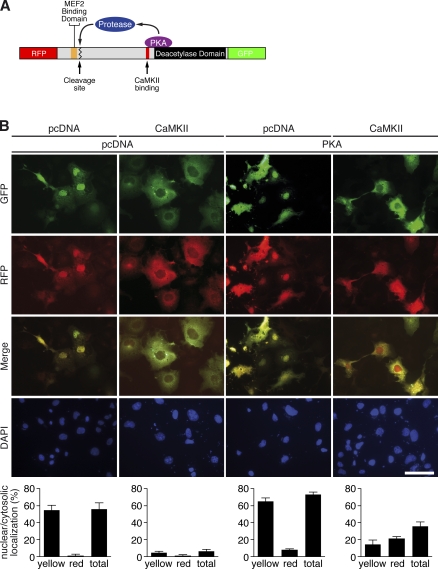
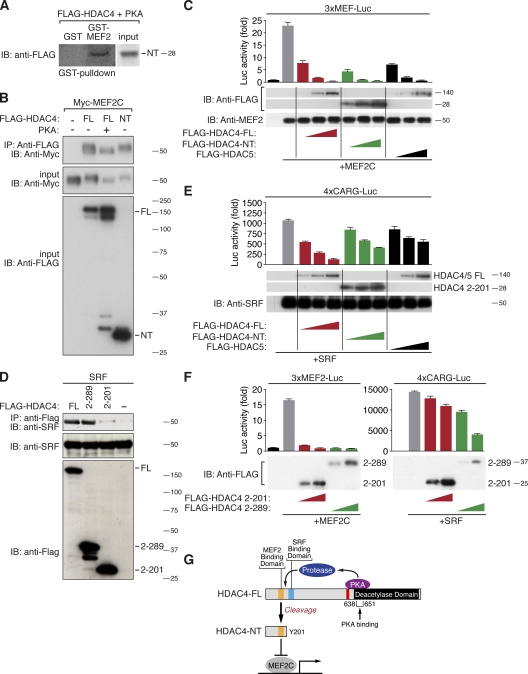
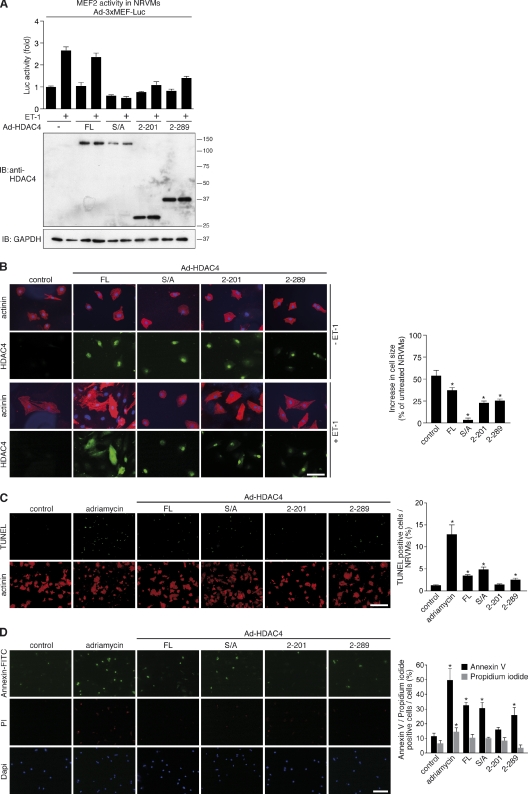
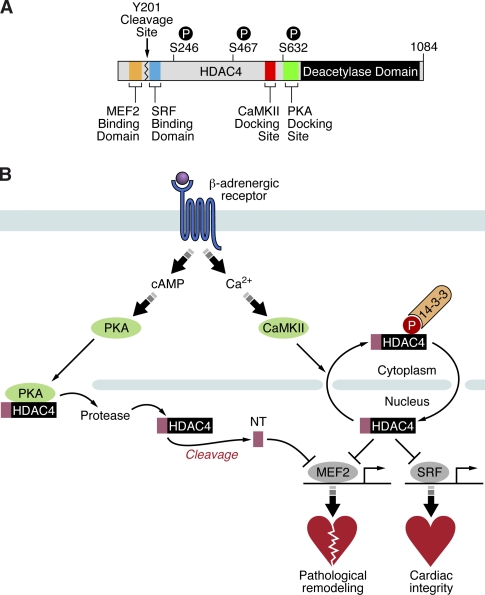
Histone deacetylase 4 (HDAC4) regulates numerous gene expression programs through its signal-dependent repression of myocyte enhancer factor 2 (MEF2) and serum response factor (SRF) transcription factors. In cardiomyocytes, calcium/calmodulin-dependent protein kinase II (CaMKII) signaling promotes hypertrophy and pathological remodeling, at least in part by phosphorylating HDAC4, with consequent stimulation of MEF2 activity. In this paper, we describe a novel mechanism whereby protein kinase A (PKA) overcomes CaMKII-mediated activation of MEF2 by regulated proteolysis of HDAC4. PKA induces the generation of an N-terminal HDAC4 cleavage product (HDAC4-NT). HDAC4-NT selectively inhibits activity of MEF2 but not SRF, thereby antagonizing the prohypertrophic actions of CaMKII signaling without affecting cardiomyocyte survival. Thus, HDAC4 functions as a molecular nexus for the antagonistic actions of the CaMKII and PKA pathways. These findings have implications for understanding the molecular basis of cardioprotection and other cellular processes in which CaMKII and PKA exert opposing effects.
Figures
References
-
- Backs J., Backs T., Neef S., Kreusser M.M., Lehmann L.H., Patrick D.M., Grueter C.E., Qi X., Richardson J.A., Hill J.A., et al. 2009. The delta isoform of CaM kinase II is required for pathological cardiac hypertrophy and remodeling after pressure overload. Proc. Natl. Acad. Sci. USA. 106:2342–2347 10.1073/pnas.0813013106 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
