

The debut of a rational treatment for an inherited neuropathy?
- PMID: 22045569
- PMCID: PMC3226011
- DOI: 10.1172/JCI60511
The debut of a rational treatment for an inherited neuropathy?
Abstract
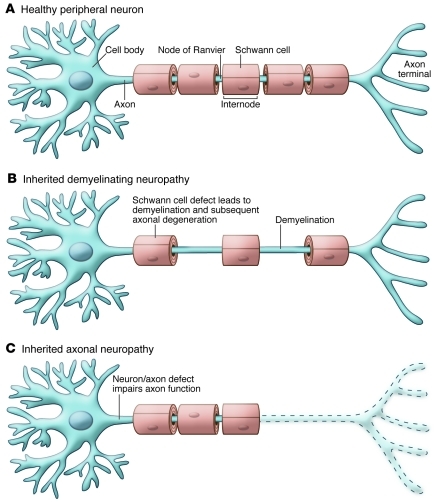
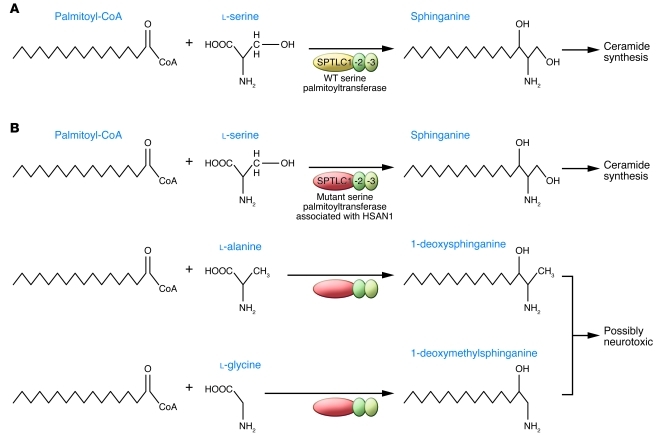
Hereditary neuropathies are common neurological conditions characterized by progressive loss of motor and/or sensory function. There are no effective treatments. Among the many causes of hereditary neuropathies are dominant mutations in serine palmitoyltransferase, long chain base subunit 1 (SPTLC1), which cause hereditary sensory and autonomic neuropathy type 1 (HSAN1). By incorporating L-alanine in place of L-serine, the mutant HSAN1–associated serine palmitoyltransferase generates deoxysphingolipids, which are thought to be neurotoxic. In this issue of the JCI, Garofalo and colleagues report that oral L-serine reverses the accumulation of deoxysphingolipids in humans with HSAN1 and in a transgenic mouse model. As oral L-serine reduces the severity of neuropathy in the mouse model of HSAN1, these data suggest a rational candidate therapy for this devastating condition.
Figures
Comment on
-
Oral L-serine supplementation reduces production of neurotoxic deoxysphingolipids in mice and humans with hereditary sensory autonomic neuropathy type 1.J Clin Invest. 2011 Dec;121(12):4735-45. doi: 10.1172/JCI57549. J Clin Invest. 2011. PMID: 22045570 Free PMC article. Clinical Trial.
References
-
- Kandel ER, Schwartz JP, Jessell TM.Principles of Neural Science . New York, New York, USA: Elsevier; 2000.
-
- Shy ME, Lupski JR, Chance PF, Klein CJ, Dyck PJ. Hereditary motor and sensory neuropathies: an overview of clinical, genetic, electrophysiologic, and pathologic features. In: Dyck PJ, Thomas PK, eds.Peripheral Neuropathy . Philadelphia, Pennsylvania, USA: Saunders; 2005:1623–1658.
-
- Timmerman V. Inherited Peripheral Neuropathies Mutation Database. Human Genome Variation Society web site. http://www.molgen.ua.ac.be/CMTMutations/. Updated February 17, 2011. Accessed September 23, 2011.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
