

A new methodology to associate SNPs with human diseases according to their pathway related context
- PMID: 22046267
- PMCID: PMC3201947
- DOI: 10.1371/journal.pone.0026277
A new methodology to associate SNPs with human diseases according to their pathway related context
Abstract
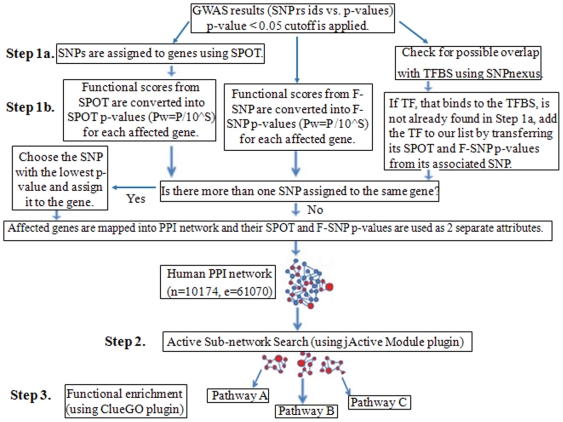
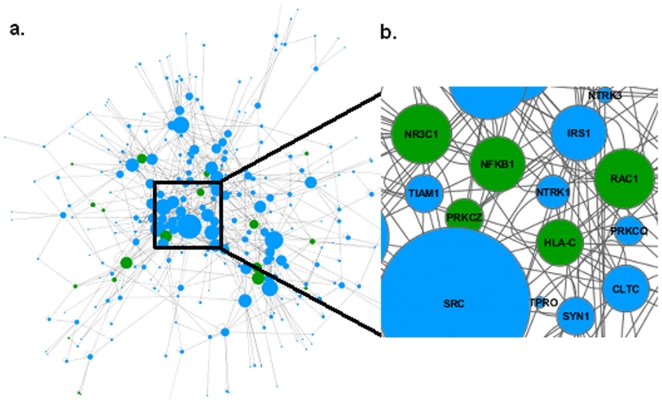
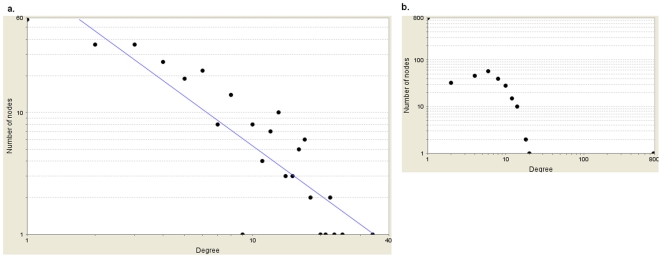
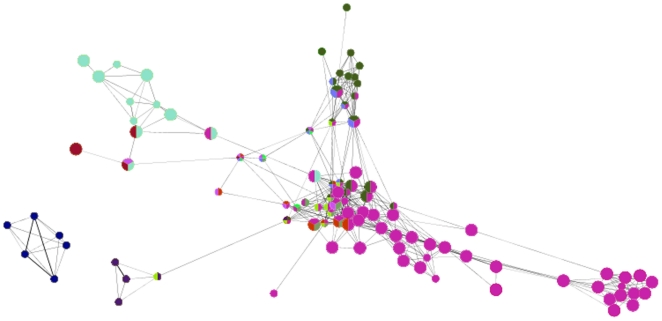
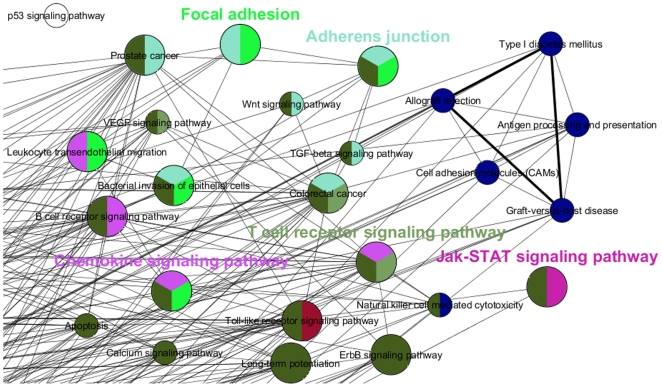
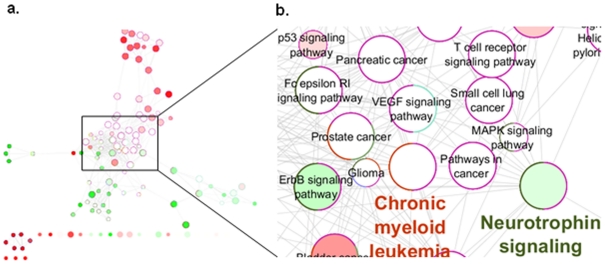
Genome-wide association studies (GWAS) with hundreds of żthousands of single nucleotide polymorphisms (SNPs) are popular strategies to reveal the genetic basis of human complex diseases. Despite many successes of GWAS, it is well recognized that new analytical approaches have to be integrated to achieve their full potential. Starting with a list of SNPs, found to be associated with disease in GWAS, here we propose a novel methodology to devise functionally important KEGG pathways through the identification of genes within these pathways, where these genes are obtained from SNP analysis. Our methodology is based on functionalization of important SNPs to identify effected genes and disease related pathways. We have tested our methodology on WTCCC Rheumatoid Arthritis (RA) dataset and identified: i) previously known RA related KEGG pathways (e.g., Toll-like receptor signaling, Jak-STAT signaling, Antigen processing, Leukocyte transendothelial migration and MAPK signaling pathways); ii) additional KEGG pathways (e.g., Pathways in cancer, Neurotrophin signaling, Chemokine signaling pathways) as associated with RA. Furthermore, these newly found pathways included genes which are targets of RA-specific drugs. Even though GWAS analysis identifies 14 out of 83 of those drug target genes; newly found functionally important KEGG pathways led to the discovery of 25 out of 83 genes, known to be used as drug targets for the treatment of RA. Among the previously known pathways, we identified additional genes associated with RA (e.g. Antigen processing and presentation, Tight junction). Importantly, within these pathways, the associations between some of these additionally found genes, such as HLA-C, HLA-G, PRKCQ, PRKCZ, TAP1, TAP2 and RA were verified by either OMIM database or by literature retrieved from the NCBI PubMed module. With the whole-genome sequencing on the horizon, we show that the full potential of GWAS can be achieved by integrating pathway and network-oriented analysis and prior knowledge from functional properties of a SNP.
Conflict of interest statement
Figures
References
-
- Elbers CC, van Eijk KR, Franke L, Mulder F, van der Schouw YT, et al. Using Genome-Wide Pathway Analysis to Unravel the Etiology of Complex Diseases. Genetic Epidemiology. 2009;33:419–431. - PubMed
-
- Altshuler D, Hirschhorn JN, Klannemark M, Lindgren CM, Vohl MC, et al. The common PPAR gamma Pro12Ala polymorphism is associated with decreased risk of type 2 diabetes. Nature Genetics. 2000;26:76–80. - PubMed
-
- Frayling TM. Genome-wide association studies provide new insights into type 2 diabetes aetiology. Nature Reviews Genetics. 2007;8:657–662. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
