

Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons
- PMID: 22047020
- PMCID: PMC3228714
- DOI: 10.1186/1471-2180-11-244
Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons
Abstract
Background: Urine within the urinary tract is commonly regarded as "sterile" in cultivation terms. Here, we present a comprehensive in-depth study of bacterial 16S rDNA sequences associated with urine from healthy females by means of culture-independent high-throughput sequencing techniques.
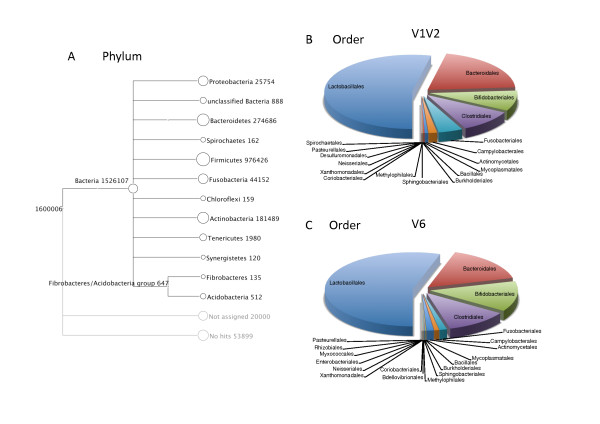
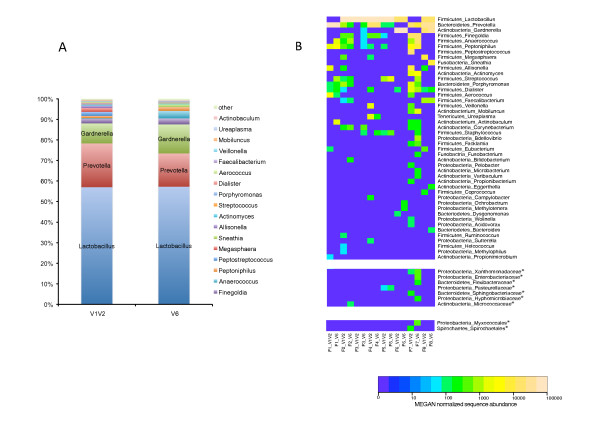
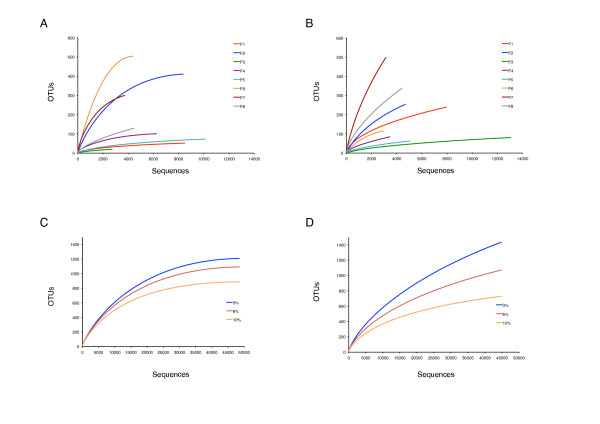
Results: Sequencing of the V1V2 and V6 regions of the 16S ribosomal RNA gene using the 454 GS FLX system was performed to characterize the possible bacterial composition in 8 culture-negative (<100,000 CFU/ml) healthy female urine specimens. Sequences were compared to 16S rRNA databases and showed significant diversity, with the predominant genera detected being Lactobacillus, Prevotella and Gardnerella. The bacterial profiles in the female urine samples studied were complex; considerable variation between individuals was observed and a common microbial signature was not evident. Notably, a significant amount of sequences belonging to bacteria with a known pathogenic potential was observed. The number of operational taxonomic units (OTUs) for individual samples varied substantially and was in the range of 20-500.
Conclusions: Normal female urine displays a noticeable and variable bacterial 16S rDNA sequence richness, which includes fastidious and anaerobic bacteria previously shown to be associated with female urogenital pathology.
Figures
References
-
- Sanz Y, Santacruz A, Gauffin P. Gut microbiota in obesity and metabolic disorders. The Proceedings of the Nutrition Society. 2010. pp. 1–8. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
