

A CFTR potentiator in patients with cystic fibrosis and the G551D mutation
- PMID: 22047557
- PMCID: PMC3230303
- DOI: 10.1056/NEJMoa1105185
A CFTR potentiator in patients with cystic fibrosis and the G551D mutation
Abstract
Background: Increasing the activity of defective cystic fibrosis transmembrane conductance regulator (CFTR) protein is a potential treatment for cystic fibrosis.
Methods: We conducted a randomized, double-blind, placebo-controlled trial to evaluate ivacaftor (VX-770), a CFTR potentiator, in subjects 12 years of age or older with cystic fibrosis and at least one G551D-CFTR mutation. Subjects were randomly assigned to receive 150 mg of ivacaftor every 12 hours (84 subjects, of whom 83 received at least one dose) or placebo (83, of whom 78 received at least one dose) for 48 weeks. The primary end point was the estimated mean change from baseline through week 24 in the percent of predicted forced expiratory volume in 1 second (FEV(1)).
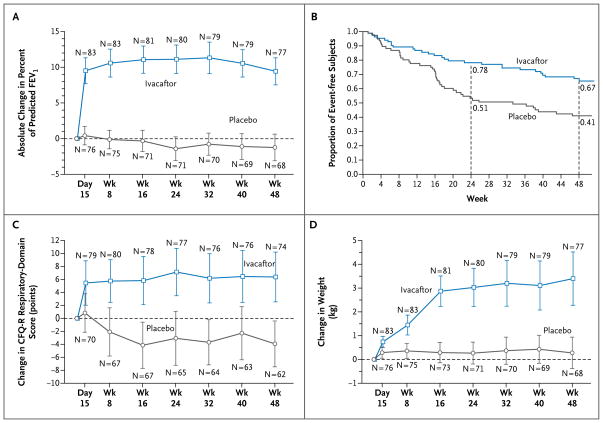
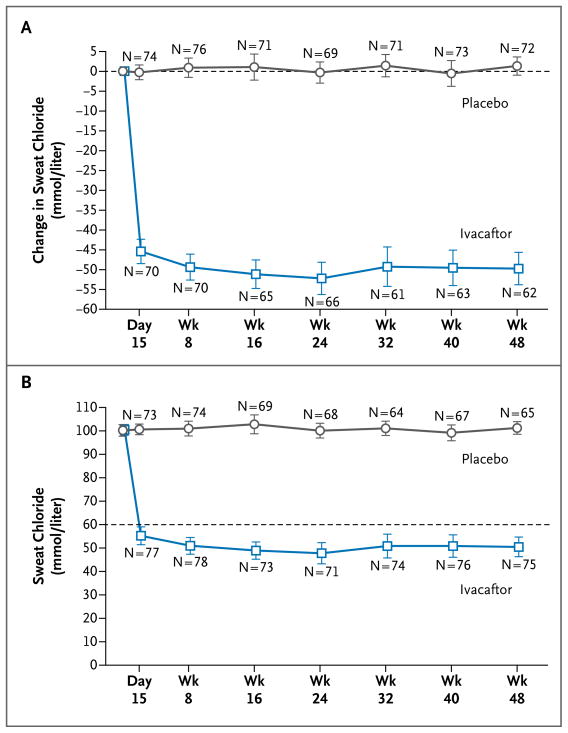
Results: The change from baseline through week 24 in the percent of predicted FEV(1) was greater by 10.6 percentage points in the ivacaftor group than in the placebo group (P<0.001). Effects on pulmonary function were noted by 2 weeks, and a significant treatment effect was maintained through week 48. Subjects receiving ivacaftor were 55% less likely to have a pulmonary exacerbation than were patients receiving placebo, through week 48 (P<0.001). In addition, through week 48, subjects in the ivacaftor group scored 8.6 points higher than did subjects in the placebo group on the respiratory-symptoms domain of the Cystic Fibrosis Questionnaire-revised instrument (a 100-point scale, with higher numbers indicating a lower effect of symptoms on the patient's quality of life) (P<0.001). By 48 weeks, patients treated with ivacaftor had gained, on average, 2.7 kg more weight than had patients receiving placebo (P<0.001). The change from baseline through week 48 in the concentration of sweat chloride, a measure of CFTR activity, with ivacaftor as compared with placebo was -48.1 mmol per liter (P<0.001). The incidence of adverse events was similar with ivacaftor and placebo, with a lower proportion of serious adverse events with ivacaftor than with placebo (24% vs. 42%).
Conclusions: Ivacaftor was associated with improvements in lung function at 2 weeks that were sustained through 48 weeks. Substantial improvements were also observed in the risk of pulmonary exacerbations, patient-reported respiratory symptoms, weight, and concentration of sweat chloride. (Funded by Vertex Pharmaceuticals and others; VX08-770-102 ClinicalTrials.gov number, NCT00909532.).
Figures
Comment in
-
Therapy for cystic fibrosis--the end of the beginning?N Engl J Med. 2011 Nov 3;365(18):1734-5. doi: 10.1056/NEJMe1110323. N Engl J Med. 2011. PMID: 22047565 No abstract available.
-
ACP Journal Club. Ivacaftor improved lung function in cystic fibrosis with G551D mutation.Ann Intern Med. 2012 Apr 17;156(8):JC4-10. doi: 10.7326/0003-4819-156-8-201204170-02010. Ann Intern Med. 2012. PMID: 22508750 No abstract available.
References
-
- Patient registry: 2008 annual data report to the Center directors. Bethesda, MD: Cystic Fibrosis Foundation; 2009.
-
- Farrell PM. The prevalence of cystic fibrosis in the European Union. J Cyst Fibros. 2008;7:450–3. - PubMed
-
- Gibson RL, Burns JL, Ramsey BW. Pathophysiology and management of pulmonary infections in cystic fibrosis. Am J Respir Crit Care Med. 2003;168:918–51. - PubMed
-
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. - PubMed
-
- Welsh MJ, Ramsey BW, Accurso FJ, Cutting GR. Cystic fibrosis. In: Scriver CR, Beaudet AR, Sly W, Valle D, editors. The metabolic and molecular bases of inherited disease. 8. New York: McGraw-Hill; 2001. pp. 521–88.
Publication types
MeSH terms
Substances
Associated data
Grants and funding
- UL1 RR024989/RR/NCRR NIH HHS/United States
- UL1 RR025005/RR/NCRR NIH HHS/United States
- UL1 RR 025005/RR/NCRR NIH HHS/United States
- P30 DK089507/DK/NIDDK NIH HHS/United States
- 1UL1 RR025744/RR/NCRR NIH HHS/United States
- P30 DK027651/DK/NIDDK NIH HHS/United States
- P30 DK27651/DK/NIDDK NIH HHS/United States
- UL1 RR025777/RR/NCRR NIH HHS/United States
- UL1 RR025014/RR/NCRR NIH HHS/United States
- K23 DK075788/DK/NIDDK NIH HHS/United States
- UL1 RR025758/RR/NCRR NIH HHS/United States
- UL1 RR 025758/RR/NCRR NIH HHS/United States
- UL1 RR024134/RR/NCRR NIH HHS/United States
- P30 DK072482/DK/NIDDK NIH HHS/United States
- UL1 RR024153/RR/NCRR NIH HHS/United States
- R01 HL105487/HL/NHLBI NIH HHS/United States
- UL1 RR025744/RR/NCRR NIH HHS/United States
- UL1-RR-024134/RR/NCRR NIH HHS/United States
- 5UL1 RR025777/RR/NCRR NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases