

Autocrine CSF-1 and CSF-1 receptor coexpression promotes renal cell carcinoma growth
- PMID: 22052465
- PMCID: PMC3251714
- DOI: 10.1158/0008-5472.CAN-11-1232
Autocrine CSF-1 and CSF-1 receptor coexpression promotes renal cell carcinoma growth
Abstract
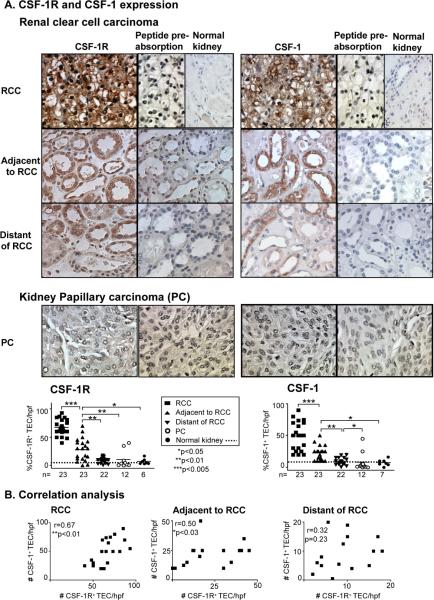
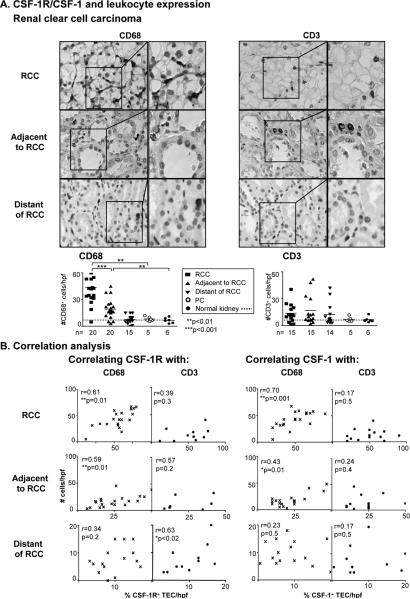
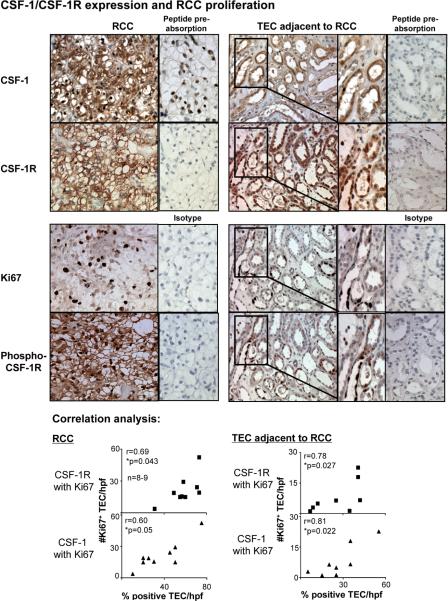
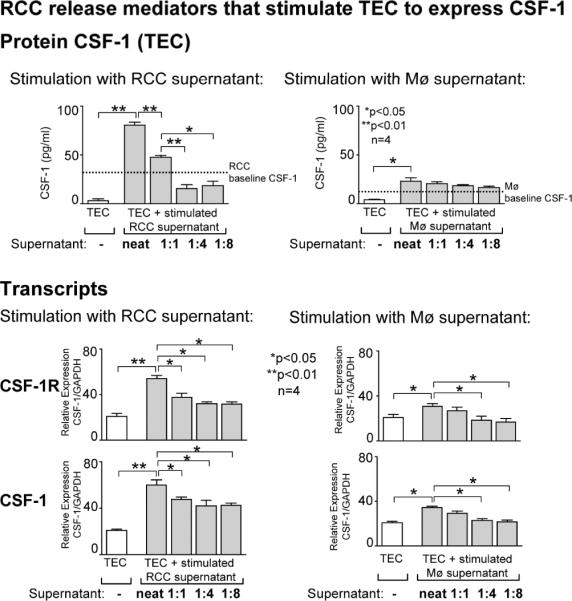
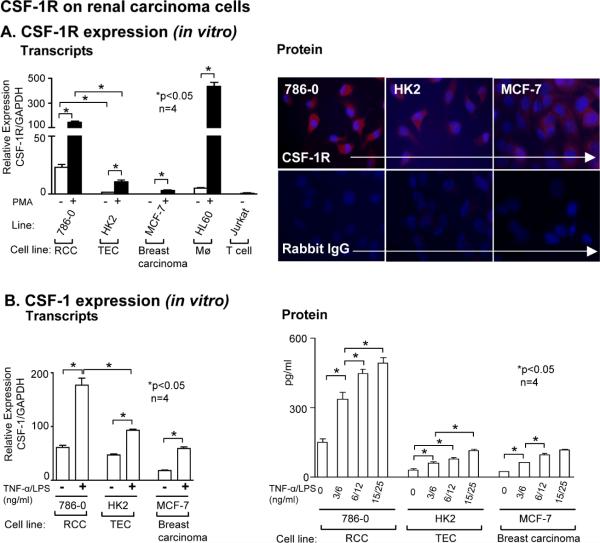
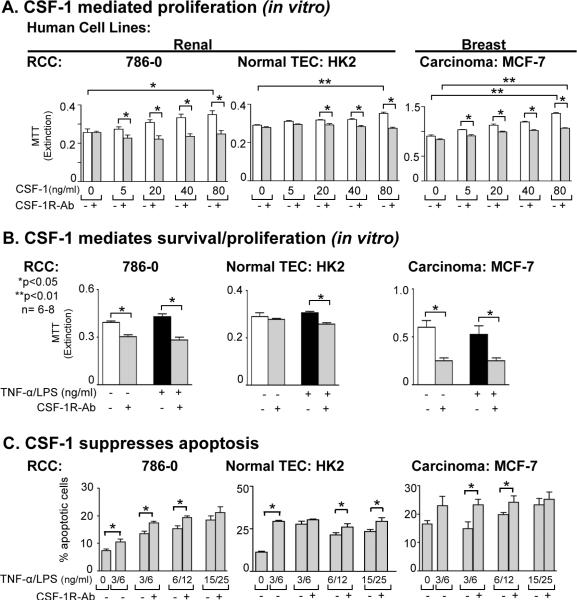
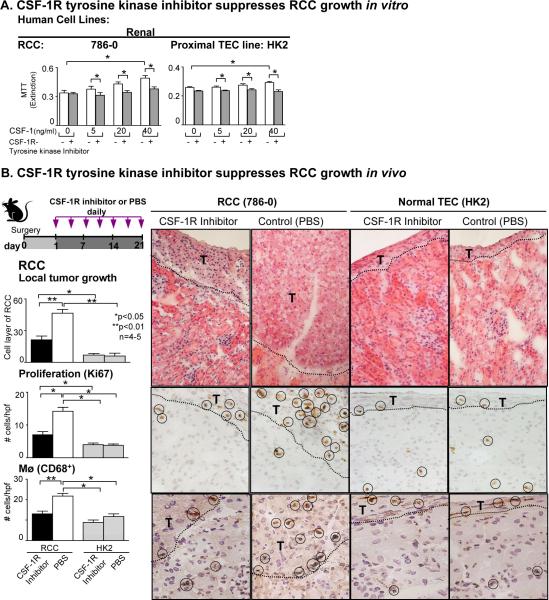
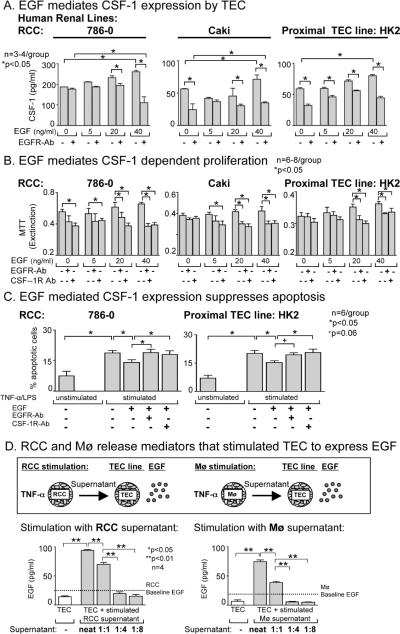
Renal cell carcinoma is increasing in incidence but the molecular mechanisms regulating its growth remain elusive. Coexpression of the monocytic growth factor colony-stimulating factor (CSF)-1 and its receptor CSF-1R on renal tubular epithelial cells (TEC) will promote proliferation and antiapoptosis during regeneration of renal tubules. Here, we show that a CSF-1-dependent autocrine pathway is also responsible for the growth of renal cell carcinoma (RCC). CSF-1 and CSF-1R were coexpressed in RCCs and TECs proximally adjacent to RCCs. CSF-1 engagement of CSF-1R promoted RCC survival and proliferation and reduced apoptosis, in support of the likelihood that CSF-1R effector signals mediate RCC growth. In vivo CSF-1R blockade using a CSF-1R tyrosine kinase inhibitor decreased RCC proliferation and macrophage infiltration in a manner associated with a dramatic reduction in tumor mass. Further mechanistic investigations linked CSF-1 and epidermal growth factor signaling in RCCs. Taken together, our results suggest that budding RCC stimulates the proximal adjacent microenvironment in the kidney to release mediators of CSF-1, CSF-1R, and epidermal growth factor expression in RCCs. Furthermore, our findings imply that targeting CSF-1/CSF-1R signaling may be therapeutically effective in RCCs.
©2011 AACR.
Figures
References
-
- Cohen HT, McGovern FJ. Renal-cell carcinoma. The New Engl J Med. 2005;353(23):2477–90. - PubMed
-
- Kovacs G, Akhtar M, Beckwith BJ, Bugert P, Cooper CS, Delahunt B, et al. The Heidelberg classification of renal cell tumours. The Journal of pathology. 1997;183(2):131–3. - PubMed
-
- Gossage L, Eisen T. Alterations in VHL as potential biomarkers in renal-cell carcinoma. Nature reviews. 7(5):277–88. - PubMed
-
- Soares MJ, Pinto M, Henrique R, Vieira J, Cerveira N, Peixoto A, et al. CSF1R copy number changes, point mutations, and RNA and protein overexpression in renal cell carcinomas. Mod Pathol. 2009;22(6):744–52. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
