

Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture
- PMID: 22052944
- PMCID: PMC3291643
- DOI: 10.1242/dmm.008409
Model for long QT syndrome type 2 using human iPS cells demonstrates arrhythmogenic characteristics in cell culture
Abstract
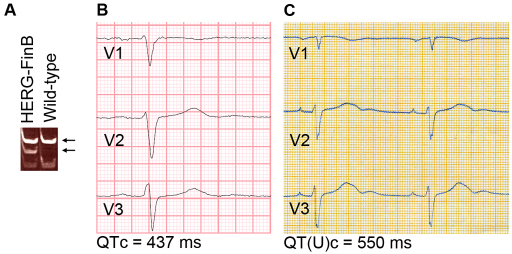
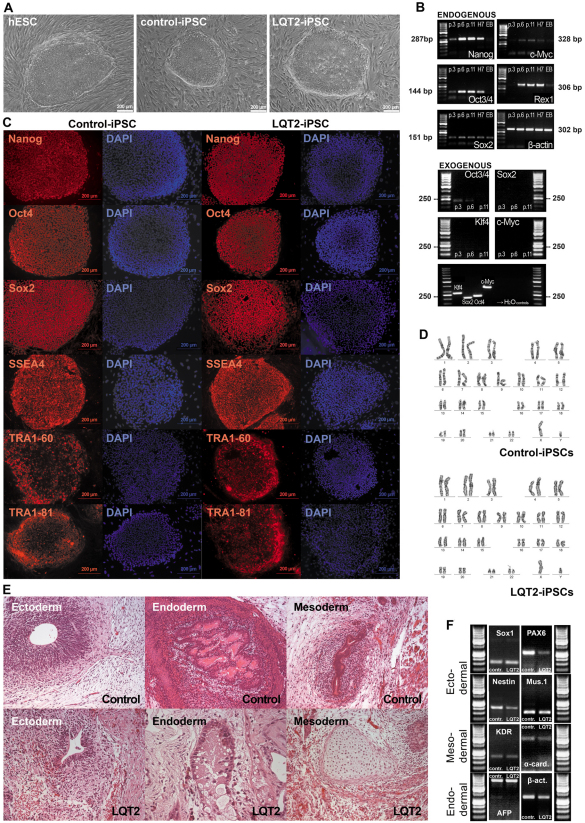
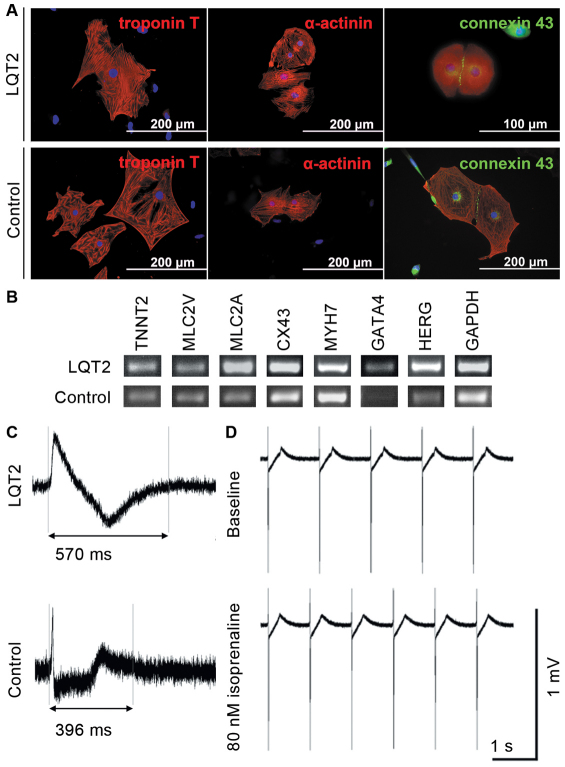
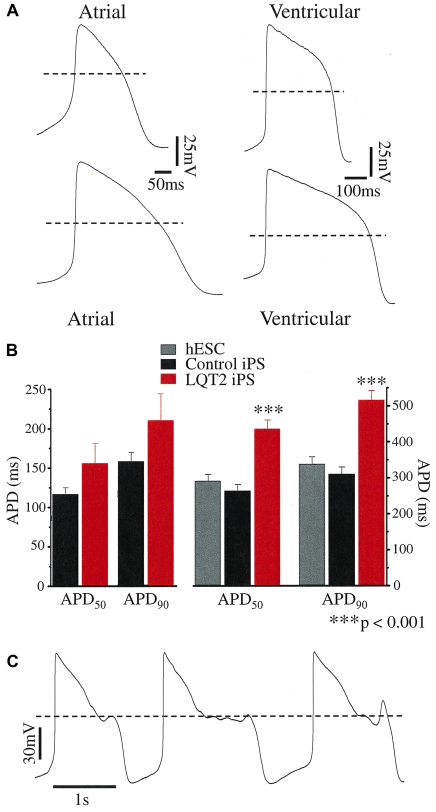
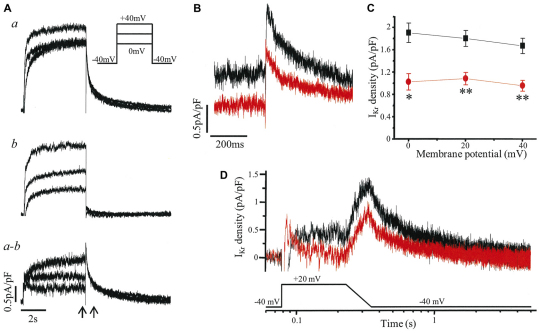
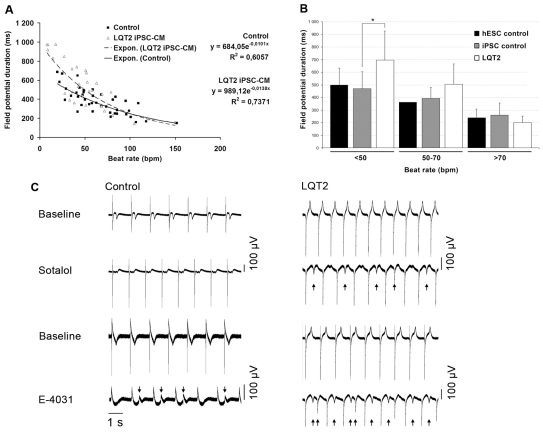
Long QT syndrome (LQTS) is caused by functional alterations in cardiac ion channels and is associated with prolonged cardiac repolarization time and increased risk of ventricular arrhythmias. Inherited type 2 LQTS (LQT2) and drug-induced LQTS both result from altered function of the hERG channel. We investigated whether the electrophysiological characteristics of LQT2 can be recapitulated in vitro using induced pluripotent stem cell (iPSC) technology. Spontaneously beating cardiomyocytes were differentiated from two iPSC lines derived from an individual with LQT2 carrying the R176W mutation in the KCNH2 (HERG) gene. The individual had been asymptomatic except for occasional palpitations, but his sister and father had died suddenly at an early age. Electrophysiological properties of LQT2-specific cardiomyocytes were studied using microelectrode array and patch-clamp, and were compared with those of cardiomyocytes derived from control cells. The action potential duration of LQT2-specific cardiomyocytes was significantly longer than that of control cardiomyocytes, and the rapid delayed potassium channel (I(Kr)) density of the LQT2 cardiomyocytes was significantly reduced. Additionally, LQT2-derived cardiac cells were more sensitive than controls to potentially arrhythmogenic drugs, including sotalol, and demonstrated arrhythmogenic electrical activity. Consistent with clinical observations, the LQT2 cardiomyocytes demonstrated a more pronounced inverse correlation between the beating rate and repolarization time compared with control cells. Prolonged action potential is present in LQT2-specific cardiomyocytes derived from a mutation carrier and arrhythmias can be triggered by a commonly used drug. Thus, the iPSC-derived, disease-specific cardiomyocytes could serve as an important platform to study pathophysiological mechanisms and drug sensitivity in LQT2.
Figures
References
-
- Ackerman M. J., Tester D. J., Jones G. S., Will M. L., Burrow C. R., Curran M. E. (2003). Ethnic differences in cardiac potassium channel variants: implications for genetic susceptibility to sudden cardiac death and genetic testing for congenital long QT syndrome. Mayo Clin. Proc. 78, 1479–1487 - PubMed
-
- Caspi O., Itzhaki I., Arbel G., Kehat I., Gepstien A., Huber I., Satin J., Gepstein L. (2008). In vitro electrophysiological drug testing using human embryonic stem cell derived cardiomyocytes. Stem Cells Dev. 18, 161–172 - PubMed
-
- Chiang C.-E., Roden D. M. (2000). The long QT syndromes: genetic basis and clinical implications. J. Am. Coll. Cardiol. 36, 1–12 - PubMed
-
- Couderc J.-P., Kaab S., Hinterseer M., McNitt S., Xia X., Fossa A., Beckmann B. M., Polonsky S., Zareba W. (2009). Baseline values and sotalol-induced changes of ventricular repolarization duration, heterogeneity, and instability in patients with a history of drug-induced torsades de pointes. J. Clin. Pharmacol. 49, 6–16 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
