

Molecular biology of lung cancer: clinical implications
- PMID: 22054881
- PMCID: PMC3367865
- DOI: 10.1016/j.ccm.2011.08.003
Molecular biology of lung cancer: clinical implications
Abstract
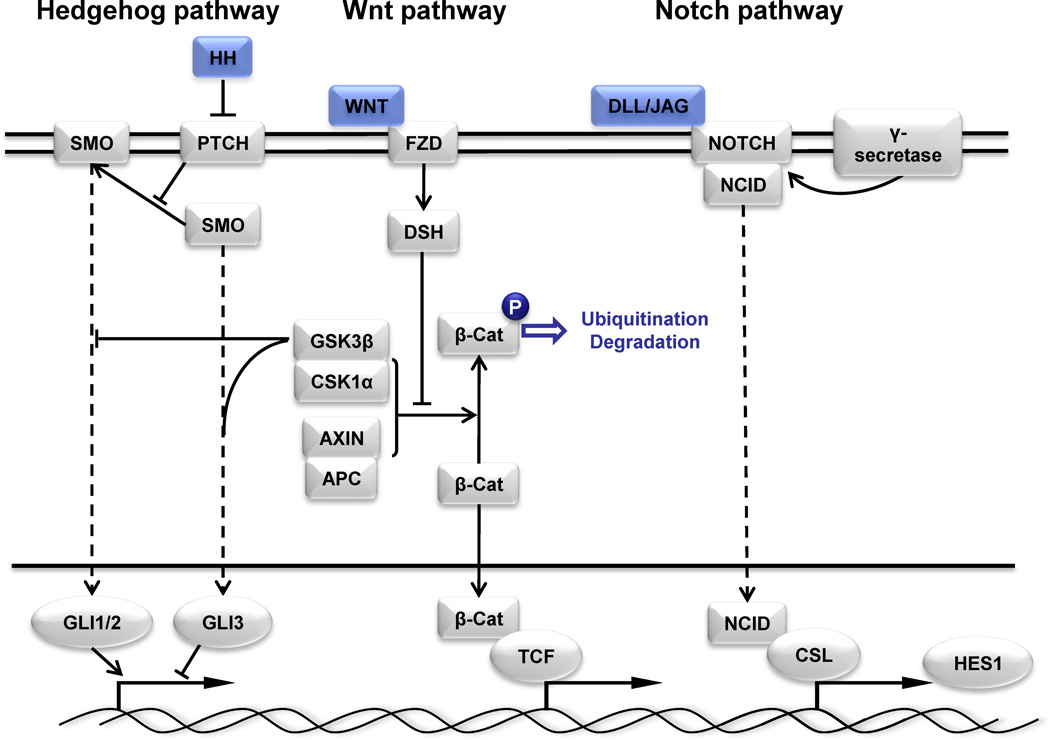
Lung cancer is a heterogeneous disease clinically, biologically, histologically, and molecularly. Understanding the molecular causes of this heterogeneity, which might reflect changes occurring in different classes of epithelial cells or different molecular changes occurring in the same target lung epithelial cells, is the focus of current research. Identifying the genes and pathways involved, determining how they relate to the biological behavior of lung cancer, and their utility as diagnostic and therapeutic targets are important basic and translational research issues. This article reviews current information on the key molecular steps in lung cancer pathogenesis, their timing, and clinical implications.
Published by Elsevier Inc.
Figures





References
-
- Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2010 Sep-Oct;60(5):277–300. - PubMed
-
- Wistuba II, Gazdar AF. Lung cancer preneoplasia. Annu Rev Pathol. 2006;1:331–348. - PubMed
-
- Sekido Y, Fong KM, Minna JD. Progress in understanding the molecular pathogenesis of human lung cancer. Biochim Biophys Acta. 1998 Aug 19;1378(1):F21–F59. - PubMed
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976 Oct 1;194(4260):23–28. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
