

A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle
- PMID: 22056929
- PMCID: PMC3256634
- DOI: 10.1128/JB.06244-11
A Rickettsia genome overrun by mobile genetic elements provides insight into the acquisition of genes characteristic of an obligate intracellular lifestyle
Abstract
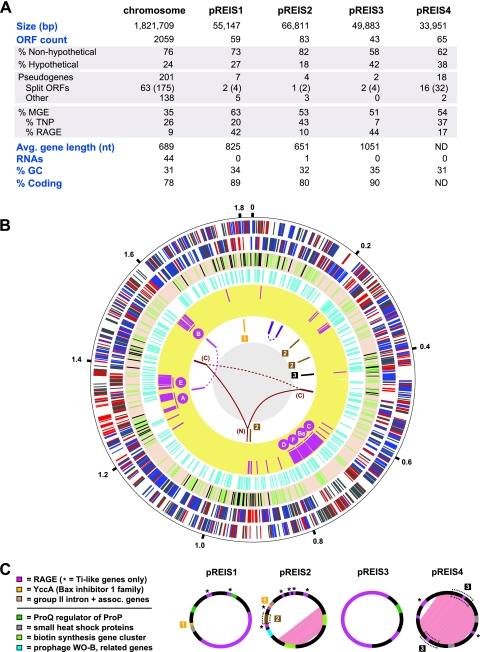
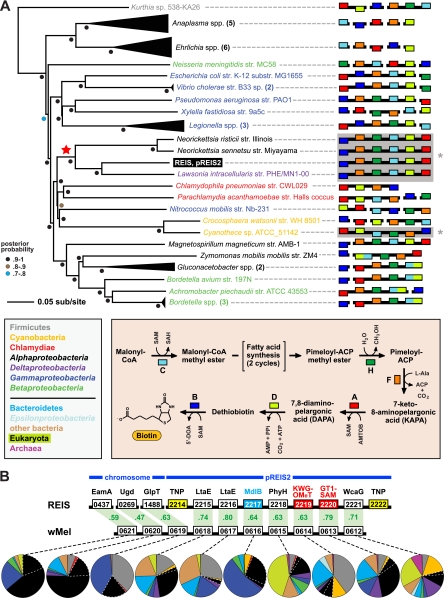
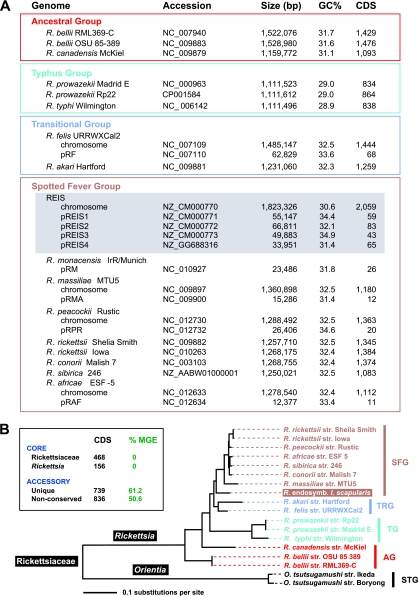
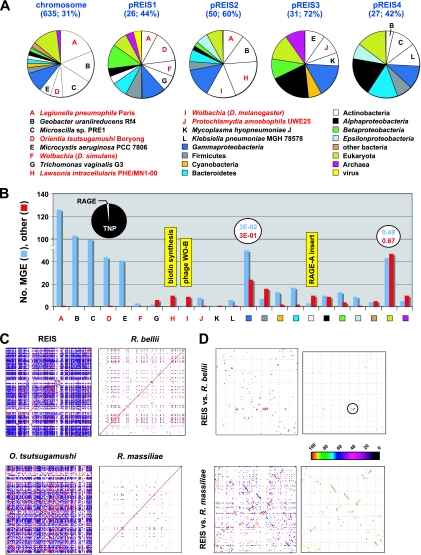
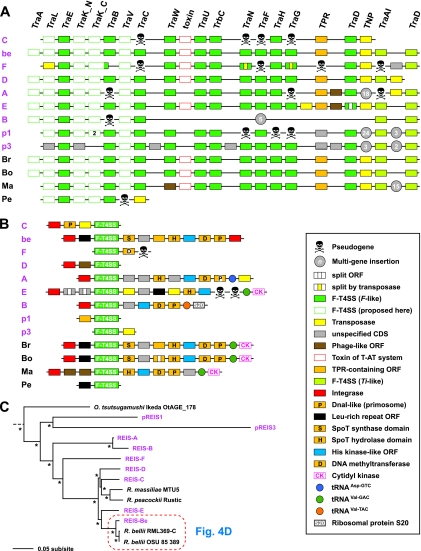
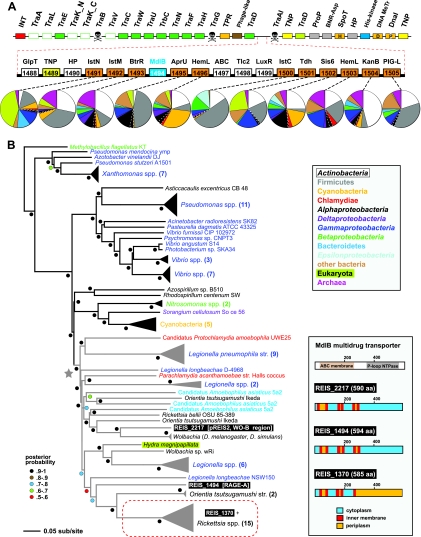
We present the draft genome for the Rickettsia endosymbiont of Ixodes scapularis (REIS), a symbiont of the deer tick vector of Lyme disease in North America. Among Rickettsia species (Alphaproteobacteria: Rickettsiales), REIS has the largest genome sequenced to date (>2 Mb) and contains 2,309 genes across the chromosome and four plasmids (pREIS1 to pREIS4). The most remarkable finding within the REIS genome is the extraordinary proliferation of mobile genetic elements (MGEs), which contributes to a limited synteny with other Rickettsia genomes. In particular, an integrative conjugative element named RAGE (for Rickettsiales amplified genetic element), previously identified in scrub typhus rickettsiae (Orientia tsutsugamushi) genomes, is present on both the REIS chromosome and plasmids. Unlike the pseudogene-laden RAGEs of O. tsutsugamushi, REIS encodes nine conserved RAGEs that include F-like type IV secretion systems similar to that of the tra genes encoded in the Rickettsia bellii and R. massiliae genomes. An unparalleled abundance of encoded transposases (>650) relative to genome size, together with the RAGEs and other MGEs, comprise ~35% of the total genome, making REIS one of the most plastic and repetitive bacterial genomes sequenced to date. We present evidence that conserved rickettsial genes associated with an intracellular lifestyle were acquired via MGEs, especially the RAGE, through a continuum of genomic invasions. Robust phylogeny estimation suggests REIS is ancestral to the virulent spotted fever group of rickettsiae. As REIS is not known to invade vertebrate cells and has no known pathogenic effects on I. scapularis, its genome sequence provides insight on the origin of mechanisms of rickettsial pathogenicity.
Figures
References
-
- Andersson SG, Stothard DR, Fuerst P, Kurland CG. 1999. Molecular phylogeny and rearrangement of rRNA genes in Rickettsia species. Mol. Biol. Evol. 16: 987–995 - PubMed
-
- Andersson SG, et al. 1998. The genome sequence of Rickettsia prowazekii and the origin of mitochondria. Nature 396: 133–140 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
