

Exploration of signals of positive selection derived from genotype-based human genome scans using re-sequencing data
- PMID: 22057783
- PMCID: PMC3325425
- DOI: 10.1007/s00439-011-1111-9
Exploration of signals of positive selection derived from genotype-based human genome scans using re-sequencing data
Abstract
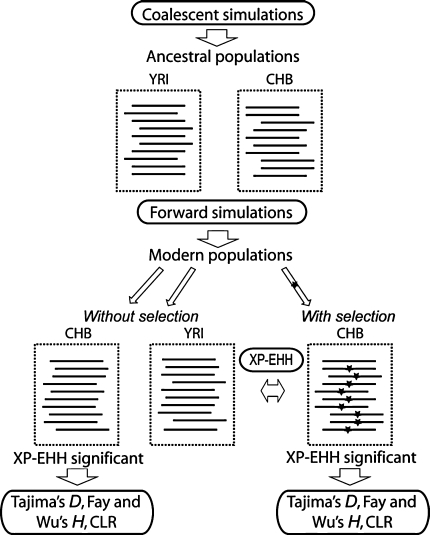
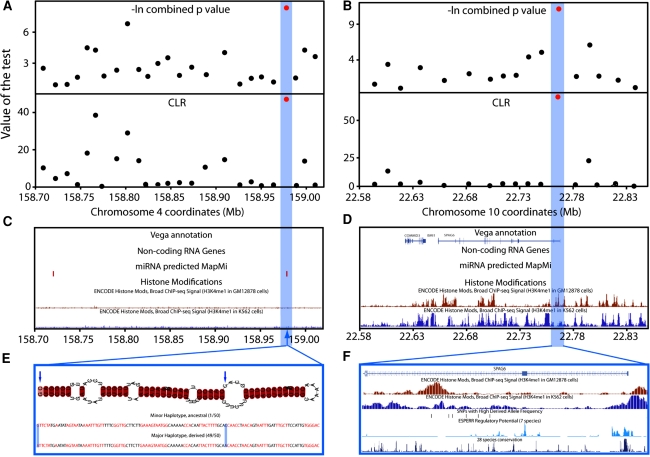
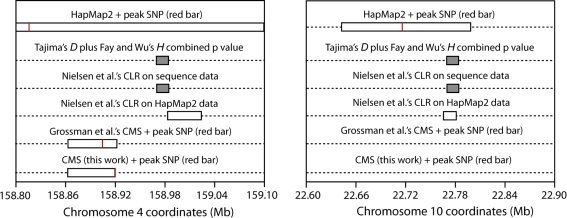
We have investigated whether regions of the genome showing signs of positive selection in scans based on haplotype structure also show evidence of positive selection when sequence-based tests are applied, whether the target of selection can be localized more precisely, and whether such extra evidence can lead to increased biological insights. We used two tools: simulations under neutrality or selection, and experimental investigation of two regions identified by the HapMap2 project as putatively selected in human populations. Simulations suggested that neutral and selected regions should be readily distinguished and that it should be possible to localize the selected variant to within 40 kb at least half of the time. Re-sequencing of two ~300 kb regions (chr4:158Mb and chr10:22Mb) lacking known targets of selection in HapMap CHB individuals provided strong evidence for positive selection within each and suggested the micro-RNA gene hsa-miR-548c as the best candidate target in one region, and changes in regulation of the sperm protein gene SPAG6 in the other.
Figures

References
-
- Cavalli-Sforza LL, Menozzi P, Piazza A. The history and geography of human genes. Princeton: Princeton University Press; 1994.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
