

Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease
- PMID: 22059643
- PMCID: PMC3223498
- DOI: 10.1186/1750-1172-6-72
Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease
Abstract
Mucopolysaccharidosis type II (MPS II) is a rare, life-limiting, X-linked recessive disease characterised by deficiency of the lysosomal enzyme iduronate-2-sulfatase. Consequent accumulation of glycosaminoglycans leads to pathological changes in multiple body systems. Age at onset, signs and symptoms, and disease progression are heterogeneous, and patients may present with many different manifestations to a wide range of specialists. Expertise in diagnosing and managing MPS II varies widely between countries, and substantial delays between disease onset and diagnosis can occur. In recent years, disease-specific treatments such as enzyme replacement therapy and stem cell transplantation have helped to address the underlying enzyme deficiency in patients with MPS II. However, the multisystem nature of this disorder and the irreversibility of some manifestations mean that most patients require substantial medical support from many different specialists, even if they are receiving treatment. This article presents an overview of how to recognise, diagnose, and care for patients with MPS II. Particular focus is given to the multidisciplinary nature of patient management, which requires input from paediatricians, specialist nurses, otorhinolaryngologists, orthopaedic surgeons, ophthalmologists, cardiologists, pneumologists, anaesthesiologists, neurologists, physiotherapists, occupational therapists, speech therapists, psychologists, social workers, homecare companies and patient societies.
Take-home message: Expertise in recognising and treating patients with MPS II varies widely between countries. This article presents pan-European recommendations for the diagnosis and management of this life-limiting disease.
Figures

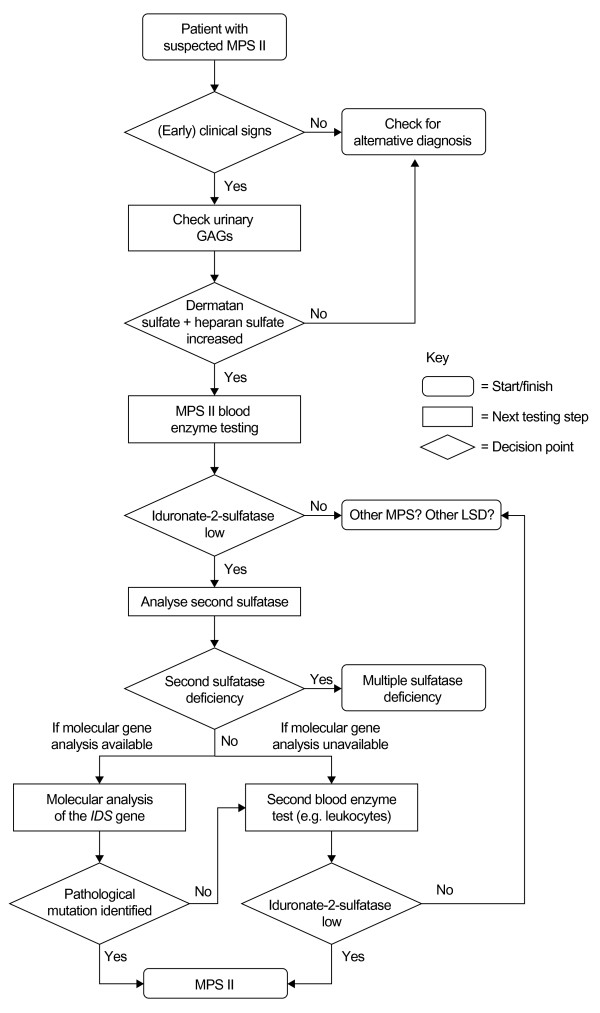
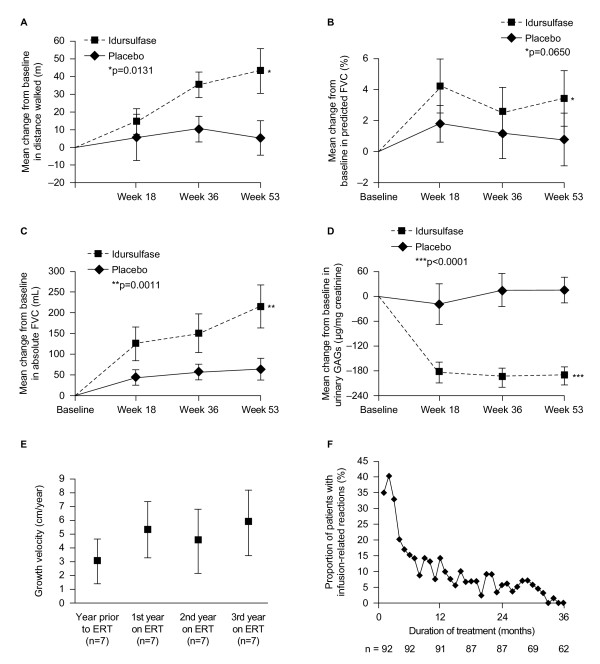
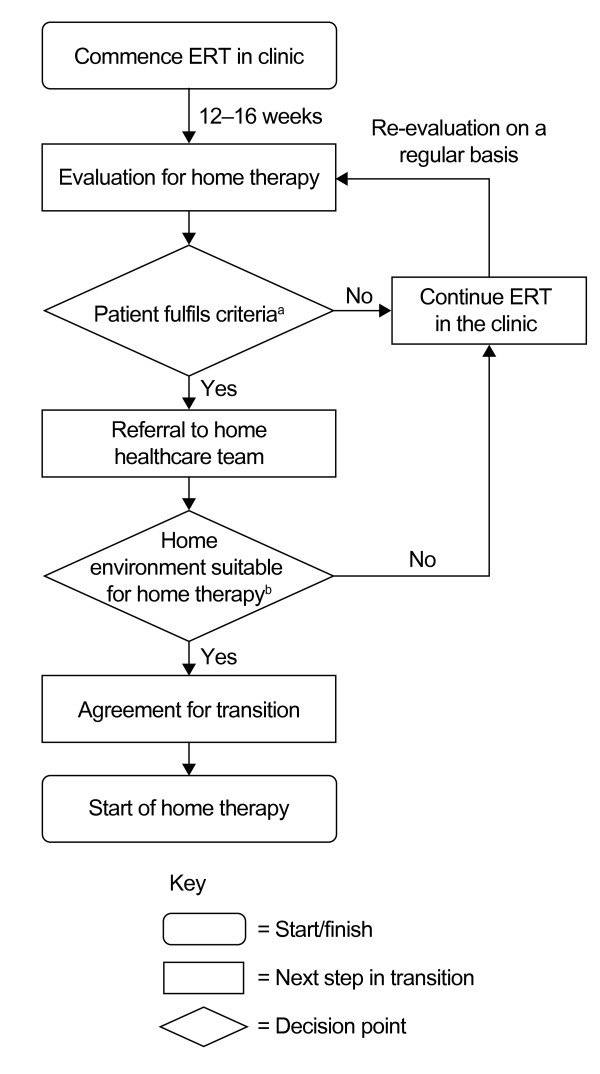
References
-
- Neufeld EF, Muenzer J. In: The metabolic and molecular bases of inherited disease. Scriver CR, Beaudet AL, Sly WS, Valle D, editor. New York: McGraw-Hill; 2001. The mucopolysaccharidoses; pp. 3421–3452.
-
- Wraith JE, Scarpa M, Beck M, Bodamer OA, De Meirleir L, Guffon N, Meldgaard Lund A, Malm G, van der Ploeg AT, Zeman J. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008;167(3):267–277. doi: 10.1007/s00431-007-0635-4. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
