

Genetic and phenotypic variations of inherited retinal diseases in dogs: the power of within- and across-breed studies
- PMID: 22065099
- PMCID: PMC3942498
- DOI: 10.1007/s00335-011-9361-3
Genetic and phenotypic variations of inherited retinal diseases in dogs: the power of within- and across-breed studies
Abstract
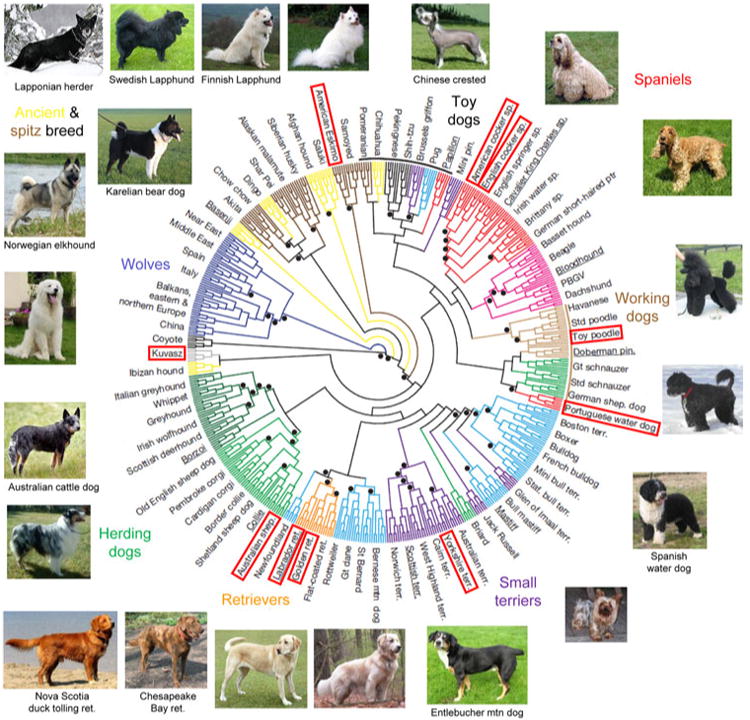
Considerable clinical and molecular variations have been known in retinal blinding diseases in man and also in dogs. Different forms of retinal diseases occur in specific breed(s) caused by mutations segregating within each isolated breeding population. While molecular studies to find genes and mutations underlying retinal diseases in dogs have benefited largely from the phenotypic and genetic uniformity within a breed, within- and across-breed variations have often played a key role in elucidating the molecular basis. The increasing knowledge of phenotypic, allelic, and genetic heterogeneities in canine retinal degeneration has shown that the overall picture is rather more complicated than initially thought. Over the past 20 years, various approaches have been developed and tested to search for genes and mutations underlying genetic traits in dogs, depending on the availability of genetic tools and sample resources. Candidate gene, linkage analysis, and genome-wide association studies have so far identified 24 mutations in 18 genes underlying retinal diseases in at least 58 dog breeds. Many of these genes have been associated with retinal diseases in humans, thus providing opportunities to study the role in pathogenesis and in normal vision. Application in therapeutic interventions such as gene therapy has proven successful initially in a naturally occurring dog model followed by trials in human patients. Other genes whose human homologs have not been associated with retinal diseases are potential candidates to explain equivalent human diseases and contribute to the understanding of their function in vision.
Conflict of interest statement
Figures
References
-
- Abramov I, Gordon J, Hendrickson A, Hainline L, Dobson V, LaBossiere E. The retina of the newborn human infant. Science. 1982;217:265–267. - PubMed
-
- Acland GM, Aguirre GD. Retinal degenerations in the dog: IV. Early retinal degeneration (erd) in Norwegian elkhounds. Exp Eye Res. 1987;44:491–521. - PubMed
-
- Acland GM, Aguirre GD. Oculoskeletal dysplasias in Samoyed and Labrador retriever dogs: nonallelic disorders akin to Stickler-like syndromes affecting humans. 2nd international DOGMAP meeting; Cambridge. 1995.
-
- Acland GM, Irby NL, Aguirre GD, Gross S. Sudden acquired retinal degeneration in the dog: clinical and morphologic characterization of the “silent retina” syndrome. Trans Am Coll Vet Ophthalmol. 1984;15:86–104.
-
- Acland G, Fletcher R, Gentleman S, Chader G, Aguirre G. Non-allelism of three genes (rcd1, rcd2, erd) for early-onset hereditary retinal degeneration. Exp Eye Res. 1989;49:983–998. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
