

Nanostructure of cellulose microfibrils in spruce wood
- PMID: 22065760
- PMCID: PMC3223458
- DOI: 10.1073/pnas.1108942108
Nanostructure of cellulose microfibrils in spruce wood
Abstract
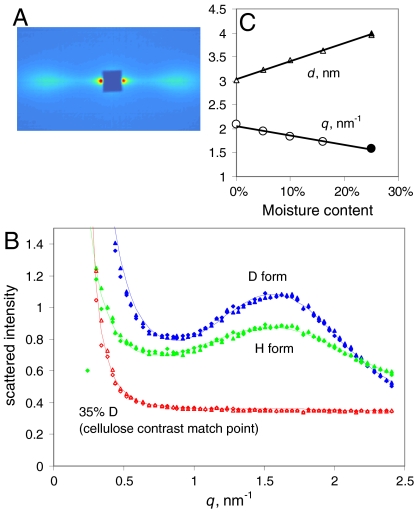
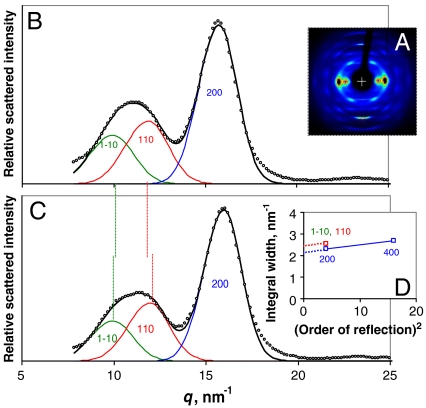
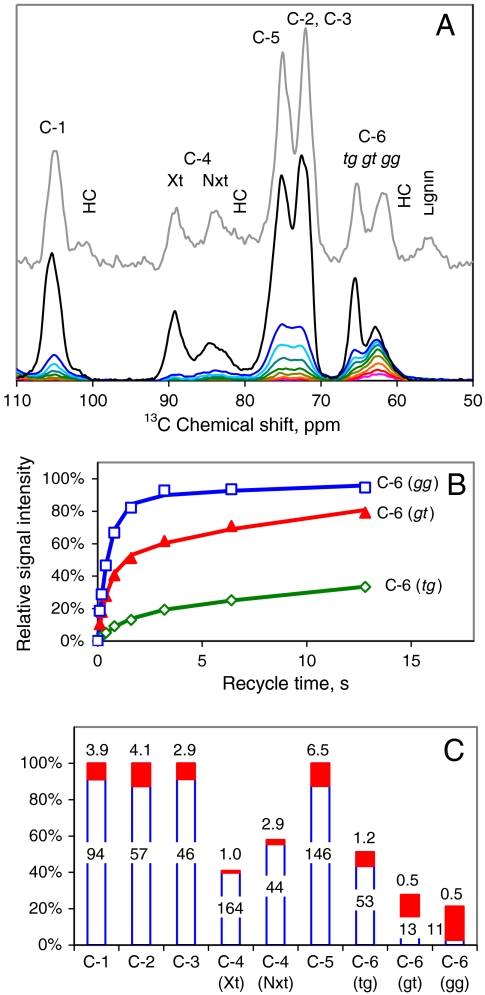
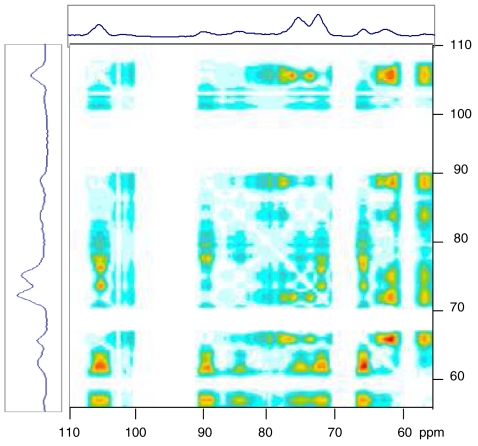
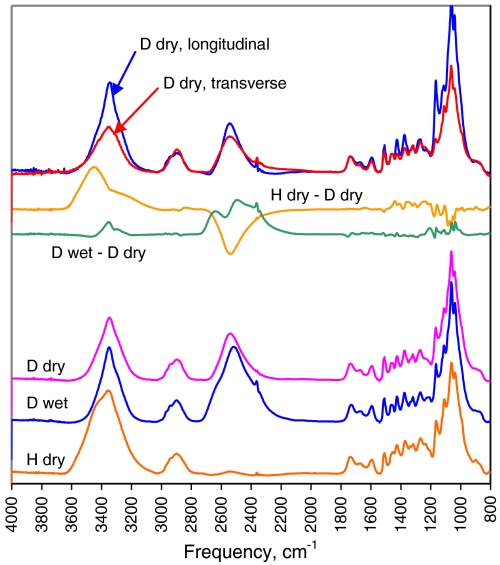
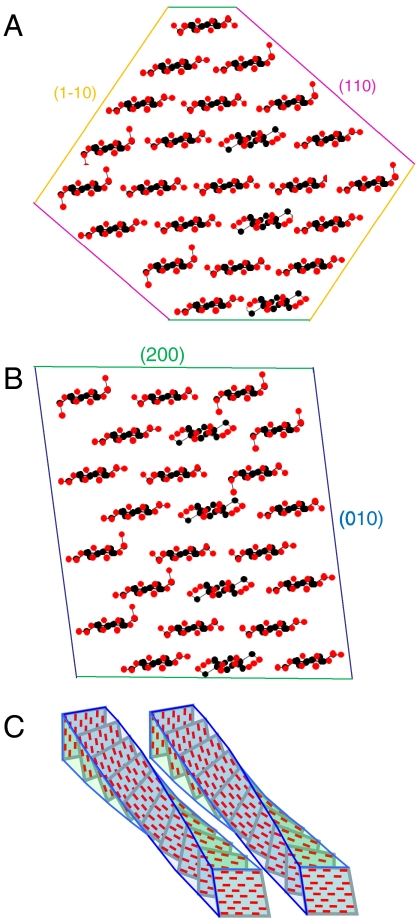
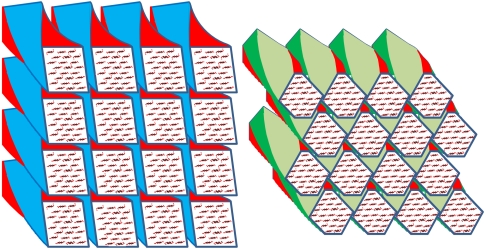
The structure of cellulose microfibrils in wood is not known in detail, despite the abundance of cellulose in woody biomass and its importance for biology, energy, and engineering. The structure of the microfibrils of spruce wood cellulose was investigated using a range of spectroscopic methods coupled to small-angle neutron and wide-angle X-ray scattering. The scattering data were consistent with 24-chain microfibrils and favored a "rectangular" model with both hydrophobic and hydrophilic surfaces exposed. Disorder in chain packing and hydrogen bonding was shown to increase outwards from the microfibril center. The extent of disorder blurred the distinction between the I alpha and I beta allomorphs. Chains at the surface were distinct in conformation, with high levels of conformational disorder at C-6, less intramolecular hydrogen bonding and more outward-directed hydrogen bonding. Axial disorder could be explained in terms of twisting of the microfibrils, with implications for their biosynthesis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Dixon RK, et al. Carbon pools and flux of global forest ecosystems. Science. 1994;263:185–190. - PubMed
-
- Carroll A, Somerville C. Cellulosic biofuels. Annu Rev Plant Biol. 2009;60:165–182. - PubMed
-
- Xu P, Donaldson LA, Gergely ZR, Staehelin LA. Dual-axis electron tomography: a new approach for investigating the spatial organization of wood cellulose microfibrils. Wood Sci Technol. 2007;41:101–116.
-
- Atalla RH, Vander Hart DL. The role of solid state C-13 NMR spectroscopy in studies of the nature of native celluloses. Solid State Nucl Mag. 1999;15:1–19. - PubMed
-
- Vietor RJ, Newman RH, Ha MA, Apperley DC, Jarvis MC. Conformational features of crystal-surface cellulose from higher plants. Plant J. 2002;30:721–731. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
