

Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation
- PMID: 22066468
- PMCID: PMC3270051
- DOI: 10.1089/ars.2011.4119
Mechanisms of altered redox regulation in neurodegenerative diseases--focus on S--glutathionylation
Abstract
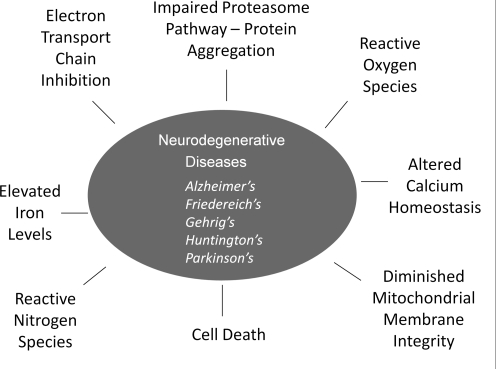
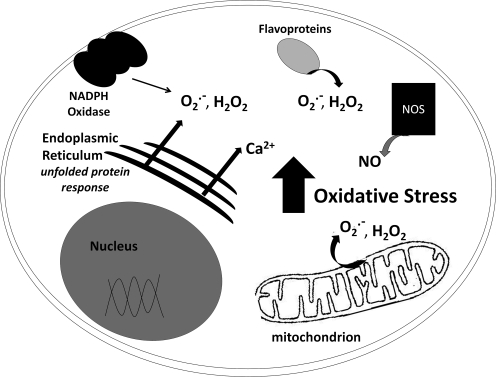
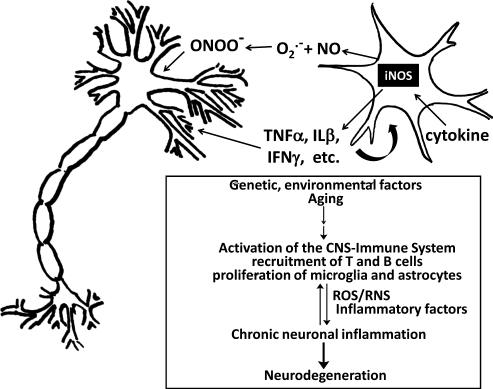
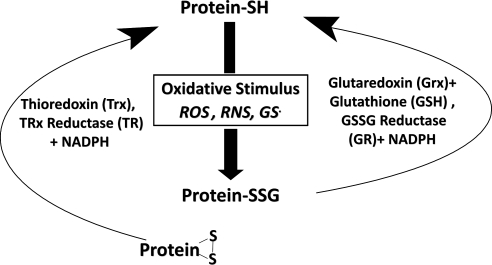
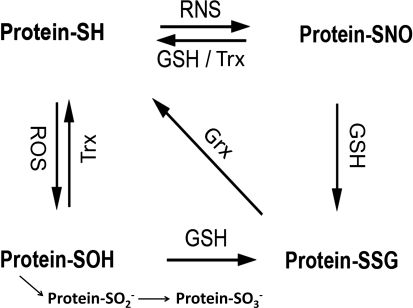
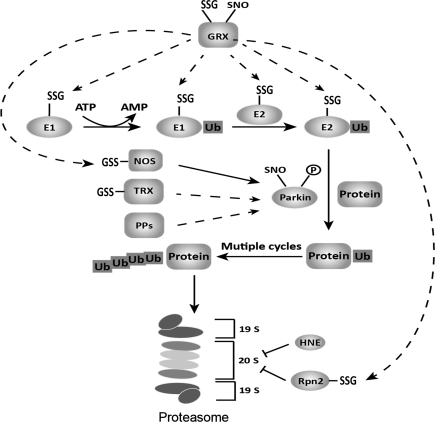
Significance: Neurodegenerative diseases are characterized by progressive loss of neurons. A common feature is oxidative stress, which arises when reactive oxygen species (ROS) and/or reactive nitrogen species (RNS) exceed amounts required for normal redox signaling. An imbalance in ROS/RNS alters functionality of cysteines and perturbs thiol-disulfide homeostasis. Many cysteine modifications may occur, but reversible protein mixed disulfides with glutathione (GSH) likely represents the common steady-state derivative due to cellular abundance of GSH and ready conversion of cysteine-sulfenic acid and S-nitrosocysteine precursors to S-glutathionylcysteine disulfides. Thus, S-glutathionylation acts in redox signal transduction and serves as a protective mechanism against irreversible cysteine oxidation. Reversal of protein-S-glutathionylation is catalyzed specifically by glutaredoxin which thereby plays a critical role in cellular regulation. This review highlights the role of oxidative modification of proteins, notably S-glutathionylation, and alterations in thiol homeostatic enzyme activities in neurodegenerative diseases, providing insights for therapeutic intervention.
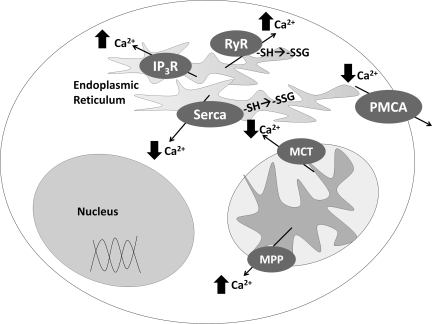
Recent advances: Recent studies show that dysregulation of redox signaling and sulfhydryl homeostasis likely contributes to onset/progression of neurodegeneration. Oxidative stress alters the thiol-disulfide status of key proteins that regulate the balance between cell survival and cell death.
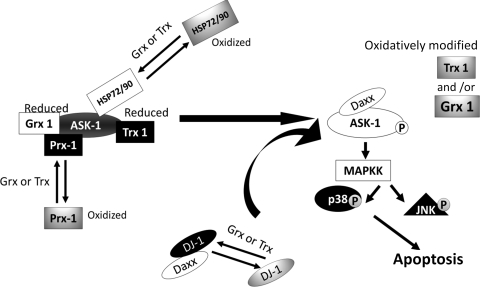
Critical issues: Much of the current information about redox modification of key enzymes and signaling intermediates has been gleaned from studies focused on oxidative stress situations other than the neurodegenerative diseases.
Future directions: The findings in other contexts are expected to apply to understanding neurodegenerative mechanisms. Identification of selectively glutathionylated proteins in a quantitative fashion will provide new insights about neuropathological consequences of this oxidative protein modification.
Figures
References
-
- Adachi T. Pimentel DR. Heibeck T. Hou X. Lee YJ. Jiang B. Ido Y. Cohen RA. S-glutathiolation of Ras mediates redox-sensitive signaling by angiotensin II in vascular smooth muscle cells. J Biol Chem. 2004;279:29857–29862. - PubMed
-
- Adachi T. Weisbrod RM. Pimentel DR. Ying J. Sharov VS. Schoneich C. Cohen RA. S-Glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004;10:1200–1207. - PubMed
-
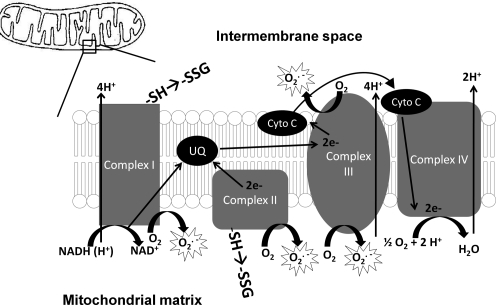
- Adam-Vizi V. Production of reactive oxygen species in brain mitochondria: Contribution by electron transport chain and non-electron transport chain sources. Antioxid Redox Signal. 2005;7:1140–1149. - PubMed
-
- Aksenov MY. Aksenova MV. Butterfield DA. Geddes JW. Markesbery WR. Protein oxidation in the brain in Alzheimer's disease. Neuroscience. 2001;103:373–383. - PubMed
-
- Akterin S. Cowburn RF. Miranda-Vizuete A. Jimenez A. Bogdanovic N. Winblad B. Cedazo-Minguez A. Involvement of glutaredoxin-1 and thioredoxin-1 in beta-amyloid toxicity and Alzheimer's disease. Cell Death Differ. 2006;13:1454–1465. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
